Chapter 6
Epigenetics and Disease

The traditional focus of human genetics has been on the ways in which alterations of DNA sequences can lead to disease (Chapter 4). In some cases, diseases and other phenotypic variation are caused by mechanisms other than changes in the DNA sequences—termed epigenetics (“upon genetics”).1 Epigenetics is the study of heritable changes in gene expression or phenotype caused by mechanisms other than changes in DNA sequences. Epigenetic modifications can cause individuals with the same DNA sequences (such as identical twins) to have different disease profiles. There are three major types of epigenetic modifications (Figure 6-1):
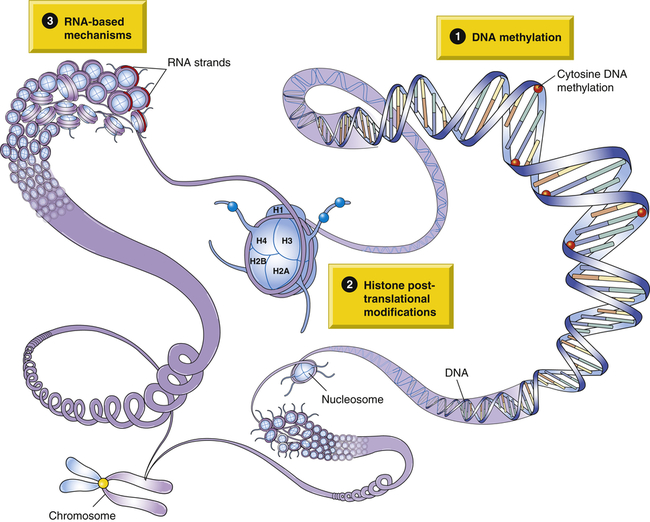
Investigators are studying the following epigenetic mechanisms: (1) DNA methylation, (2) histone modifications, and (3) RNA based-mechanisms. See text for discussion.
1. DNA methylation: The attachment of a methyl group to a cytosine base is followed by a guanine base (a “CpG dinucleotide”) (Figures 6-1 and 6-2).2 DNA methylation causes a gene to become transcriptionally inactive or silent.3 When the DNA sequence in the promoter region of a gene becomes heavily methylated, the DNA is less likely to be transcribed into mRNA. Methylation along with histone hypoacetylation (the acetyl group consists of a methyl group single-bonded to a carbonyl) and condensation of chromatin together inhibit the binding of transcription-binding factors (see Chapter 4) that regulate transcription. In other words, the gene becomes transcriptionally inactive. Aberrant methylation can lead to silencing of tumor-suppressor genes in the development of cancer (see Chapter 12 and Figure 6-1). DNA methylation is a key component of X-inactivation, the transcriptional silencing of genes on the X chromosome, discussed in Chapter 4. As with X-inactivation, epigenetic modifications are maintained in successive mitotic cell divisions, but most are erased from the genome when new gametes (sperm or egg cells) are formed.
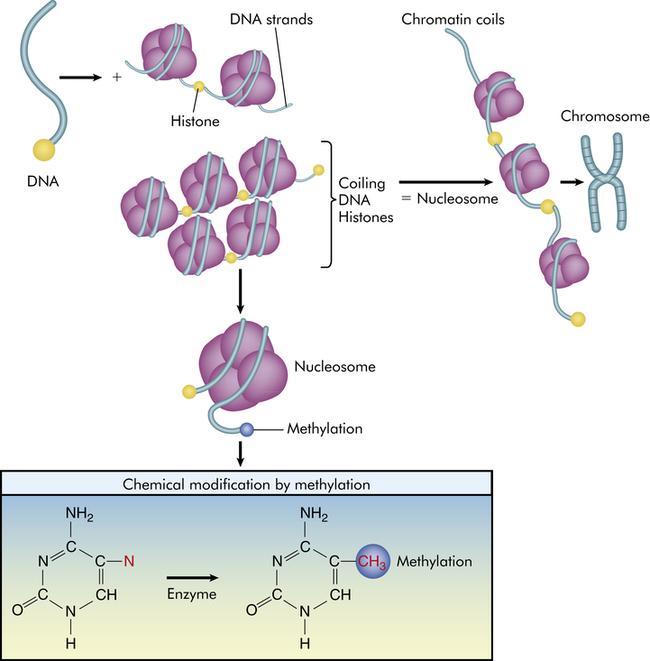
Because DNA is a long molecule, it needs packaging to fit in the tiny nucleus. Packaging involves coiling of the DNA in a “left-handed” spiral around spools, made of four pairs of proteins individually known as histones and collectively as the histone octamer. The entire spool is called a nucleosome (also see Figure 1-2). Nucleosomes are organized into chromatin, the repeating building blocks of a chromosome. Histone modifications are correlated with methylation, are reversible, and occur at multiple sites. Methylation occurs at the 5 position of cytosine and provides a “footprint” or signature as a unique epigenetic alteration (red). When genes are expressed, chromatin is open or active; however, when chromatin is condensed because of methylation and histone modification, genes are inactivated.
2. Histone modification (e.g., histone acetylation and deacetylation, alterations in chromatin): Chromatin compaction and organization help to regulate gene expression, determining and maintaining cell identity. Chromatin structure must be controlled in self-renewing and differentiated cells in cell renewal systems. For example, there are differences in chromatin structure in stem cells and terminally differentiated cells.4
3. Micro-ribonucleic acids (miRNAs or miRs): These RNAs are encoded by short DNA sequences (approximately 22 nucleotides) located in introns of genes or transcribed from noncoding DNA located between genes. Gene expression networks can be regulated by changes in miRNAs and other noncoding RNAs. Noncoding RNAs (ncRNAs) have been shown to regulate gene expression by novel mechanisms such as RNA interference, gene co-suppression, gene silencing, imprinting (see p. 187), and DNA demethylation. It is becoming clear that these novel RNAs perform critical functions during development and cell differentiation. MicroRNAs regulate diverse signaling pathways, and those that stimulate cancer development and progression are called oncomirs. For example, miRNAs have been linked to carcinogenesis because they can act as either oncogenes or tumor-suppressor genes (see Chapter 12).
Epigenetics and Development
Much remains to be learned about factors that cause epigenetic modifications. Findings so far indicate that specific environmental or nongenetic factors, such as diet and exposure to certain chemicals, can drive such modifications. For example, alcohol has been shown to affect methylation patterns in animal models,5 so the harmful effects of fetal alcohol exposure may be mediated through epigenetic mechanisms. Maternal dietary deficiency during pregnancy may cause epigenetic modifications of fetal genes, leading to an increased risk of obesity and diabetes in the offspring later in life.6 In some animal models, the insulin-like growth factor 2 gene (IGF2) is a target of these epigenetic modifications. Although the observed changes in methylation status of CpG sequences in these genes are typically small, it is possible that they affect phenotypic development. The hereditary transmission of epigenetic changes to successive generations has been termed epigenetic transgenerational inheritance. If demonstrated to occur in humans, transgenerational inheritance could have important implications for disease and disease prevention.
Twin Studies Provide Insights on Epigenetic Modification
A powerful means to test for epigenetic effects is to compare methylation and other signatures of epigenetic modification in identical (monozygotic) twin pairs, whose DNA sequences are essentially the same. As twins age, they demonstrate increasing differences in methylation patterns of the DNA sequences of their somatic cells; these changes are reflected in increasing numbers of phenotypic differences.7 Twins with significant lifestyle differences (e.g., smoking vs. nonsmoking) accumulated larger numbers of differences in their methylation patterns. The twins, despite having identical DNA sequences, become more and more different as a result of epigenetic changes, which in turn affect the expression of genes. These results, along with findings generated in animal studies, suggest that changes in epigenetic patterns may be an important part of the aging process.8
Epigenetics and Cancer
DNA Methylation and Cancer
The best evidence for epigenetic effects on disease risk comes from studies of human cancer (Figure 6-3).9,10 Tumor cells typically exhibit hypomethylation (decreased methylation), which can increase the activity of oncogenes (see Chapter 12). Hypomethylation increases as tumors progress from benign neoplasms to malignancy. In addition, the promoter regions of tumor-suppressor genes are often hypermethylated, which decreases their rate of transcription and their ability to inhibit tumor formation. Hypermethylation of the promoter region of the RB1 gene is often seen in retinoblastoma,11 and hypermethylation of the BRCA1 gene is seen in some cases of inherited breast cancer.12 Similarly, von Hippel-Lindau disease, in which renal cell carcinomas frequently occur, can be caused by hypermethylation of the VHL promoter region.
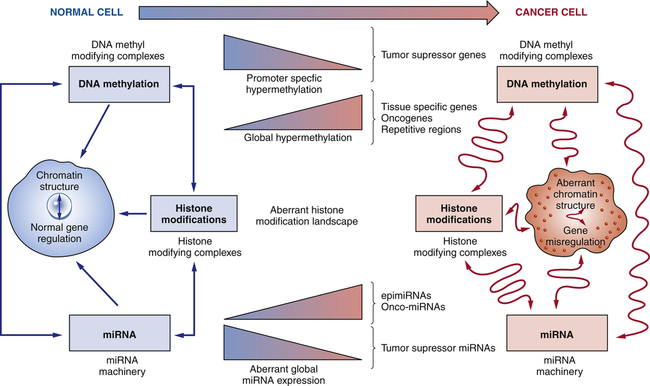
Oncogenesis involves accumulated genetic alterations combined with the epigenetic changes: DNA methylation, histone modifications, and miRNAs. In cancer cells, tumor-suppressor genes become hypermethylated and with histone modifications cause abnormal gene silencing. The gene silencing of tumor-suppressor genes results in tumor progression. Global hypomethylation leads to chromosomal instability and fragility. Additionally, these modifications create abnormal mRNA and miRNA expression, which leads to activation of oncogenes and silencing of tumor-suppressor genes. (Adapted from Sandoval J, Esteller M: Curr Opin Genet Dev 22:50–55, 2012.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


