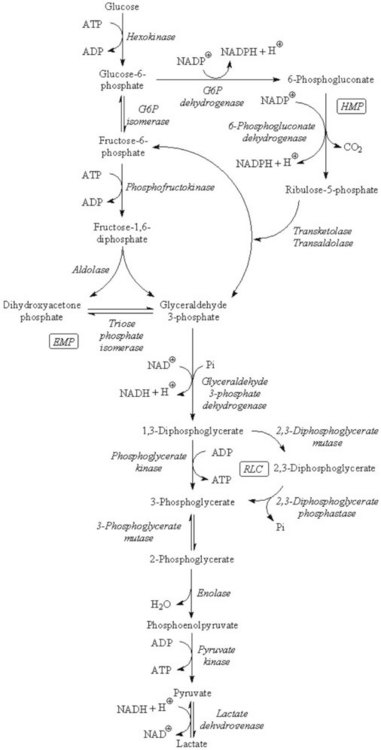
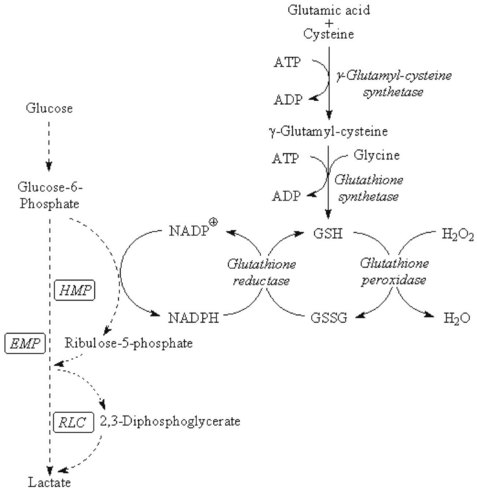
Chapter 23 Normal human red cells survive in the circulation for approximately 120 days, using energy to maintain the electrolyte gradient between plasma and red cell cytoplasm and to keep hemoglobin and the sulfhydryl groups of red cell enzymes and membrane proteins in the reduced state. Because of the absence of a nucleus and mitochondria, the red cell is incapable of generating energy via the (oxidative) Krebs cycle and depends mainly on the anaerobic conversion of glucose by the Embden-Meyerhof pathway (EMP, or direct glycolytic pathway) and the oxidative hexose monophosphate pathway (HMP, or pentose phosphate shunt) (Figure 23-1). Numerous red cell enzymes are involved in these pathways, thereby providing the cell with the necessary high-energy phosphates (primarily ATP) and reducing power (NADH, NADPH). Deficiencies of any of these red cell enzymes may result in impaired ATP generation or the inability to withstand oxidative stress and, consequently, loss of function of the erythrocyte. By far, a majority of these disorders are hereditary in nature, although acquired deficiencies have been described, mainly in malignant disorders involving the bone marrow.37 Hereditary enzymatic defects in these pathways are able to (1) disturb the erythrocyte’s integrity, (2) shorten its survival, and (3) produce hereditary nonspherocytic hemolytic anemia (HNSHA). In general, deficiencies of enzymes involved in ATP generation lead to chronic hemolytic anemia. Other enzyme deficiencies cause acute episodes of severe hemolysis [e.g., when oxidative stress on the red cell is increased (as in some types of glucose-6-phosphate dehydrogenase deficiency)]. Red cell morphology is, in general, unremarkable, except for the case of pyrimidine-5′-nucleotidase deficiency, which is characterized by prominent stippling (see later). Disorders have been described in EMP, HMP, the Rapoport-Luebering cycle, the glutathione pathway (Figure 23-2), purine-pyrimidine metabolism, and methemoglobin reduction. This section describes the clinically important red cell enzymes involved in these metabolic pathways and the disorders associated with defects in these pathways. In addition, diagnostic strategies and pitfalls of laboratory diagnostics for these enzyme deficiencies are explained. The laboratory methods described have been used for decades and are well documented. During the past few years, however, molecular diagnostics have proven to be an indispensable tool in the diagnosis of hereditary red cell enzyme deficiencies. In mammalian tissues, four isozymes of HK with different enzymatic properties exist: HK-I to III with an Mr of 100 kDa, and HK-IV (or glucokinase) with an Mr of 50 kDa. HK-I is the predominant HK isozyme in tissues that depend strongly on glucose for their physiologic functioning, such as brain, muscle, and erythrocytes. HK-I displays unique regulatory properties in its sensitivity to inhibition by physiologic concentrations of the product G6P and, moreover, relief of this inhibition by inorganic phosphate and by high concentrations of glucose.194 In addition, the enzyme depends on magnesium. HK-I is a homodimer,7,124 and elucidation of the structures of human and rat HK-I has provided substantial insight into ligand-binding sites and subsequent modes of interaction.5–7,124,164 Apart from HK-I, erythrocytes contain a specific subtype of HK: HK-R.125 Both HKs are encoded by the gene HK1, localized on chromosome 10q22 and spanning more than 130 kb.9 The structure of HK1 is complex: it encompasses 25 exons that, by tissue-specific transcription, generate multiple transcripts through alternative splicing of different 5′ exons.9 Erythroid-specific transcriptional control results in the unique red blood cell–specific mRNA that differs from HK-I mRNA at the 5′-untranslated region (5′-UTR) and at the first 63 nucleotides of the coding region.126,127,158 Consequently, HK-R lacks the porin-binding domain that mediates HK-I binding to mitochondria.127 HK deficiency (OMIM 235700)* is a rare, recessively inherited disease with chronic nonspherocytic hemolytic anemia as the predominant clinical feature. The phenotypic expression of the disease is heterogeneous, as with most glycolytic red cell enzyme deficiencies. The spectrum ranges from severe neonatal hemolysis and death to a fully compensated chronic hemolytic anemia. In general, patients benefit from splenectomy. Because HK activity is dependent on red cell age, reticulocytosis, usually present in HK-deficient patients, may obscure the enzyme deficiency. Other age-dependent red cell enzymes (e.g., pyruvate kinase, G6PD) should be measured simultaneously as an internal control to assess the influence of reticulocyte enzyme activity. The Downeast anemia mutation in mice represents an animal model of generalized HK deficiency.142 Eighteen families with HK deficiency have been described to date, and gene changes have been characterized in only four patients.32,33,56,90,186 Three amino acid substitutions in HK are known to date to affect enzyme stability (p.Leu529Ser)32,33 or the enzyme’s active site (p.Thr680Ser).186 The third missense mutation encodes an p.Arg93Gln change and causes aberrant pre-mRNA processing of the HK1 transcript.56 A lethal case of HK deficiency was found to be due to an intragenic deletion of 9.5 kb, causing the deletion of exons 5 to 8 of HK1, resulting in a null allele.90 An intriguing mutation was identified in the erythroid-specific promoter. This single NT change downregulates erythroid-specific transcription of HK1 and, hence, specifically affects HK-R production.56 G6P isomerase (GPI; EC 5.3.1.9) [also known as phosphoglucose isomerase (PGI)], catalyzes the interconversion of G6P and fructose 6-phosphate (F6P)—the second step of the EMP. As a result of this reversible reaction, products of the hexose monophosphate pathway can be recycled to G6P. Besides being a housekeeping enzyme of glycolysis, monomeric GPI is identical to neuroleukin and as such exerts lymphokine properties.101,168 In addition, autoantibodies against GPI seem to be involved in rheumatoid arthritis.159 The crystal structure of human GPI has been resolved. The enzyme is a homodimer consisting of two subunits of 63 kDa each. The dimeric form of GPI is a prerequisite for catalytic activity because the active site of the enzyme is composed of polypeptide chains from both subunits.50,145 The gene encoding GPI (GPI) is located on chromosome 19q13.1 and consists of 18 exons, spanning at least 50 kb, with a cDNA 1.9 kb in length.197 GPI deficiency (OMIM 172400) is an autosomal recessive disease and, after G6PD and PK deficiency, the third most common red cell enzymopathy. Patients are homozygous or compound heterozygous, showing mild to severe chronic hemolytic anemia. Neonatal death,80 hydrops fetalis,115,144 neurologic symptoms, and granulocyte dysfunction162 have been reported. The phenotype of homozygous GPI-deficient mice resembles that of human enzymopathy.120 GPI knockout mice die in the embryologic state.192 More than 50 families with GPI deficiency have been described worldwide.47,102,145,148,149 Most mutations are missense mutations, but nonsense and splice-site mutations have also been reported.102 Using the three-dimensional model of GPI, many of the missense mutations illustrate just how critical the precise three-dimensional structure is for correct function.145,149 Most of the mutations disrupt key interactions that contribute directly or indirectly to the active site architecture.48,145 It is intriguing to note that combined deficiency of GPI and glucose-6-phosphate dehydrogenase appears to be associated with a more favorable clinical outcome than does either deficiency alone.47 The genes encoding PFK-L (PFKL) and PFK-M (PFKM) are located on chromosomes 21q22.3 and 12q13.3, respectively. The PFKM gene spans more than 30 kb, containing 27 exons and at least 3 promoter regions.128,198 PFK deficiency is a rare autosomal, recessively inherited disorder. Because red cells contain both M and L subunits, mutations affecting either of the genes coding for these subunits will affect PFK activity. Thus, when the L subunit is affected, RBCs contain only M4 PFK homotetramers and are partially PFK deficient. In such cases, patients display a mild hemolytic disorder without myopathy. However, when the M subunit is deficient, partial PFK deficiency in red blood cells is accompanied by virtually absent PFK activity in muscle. Consequently, a deficiency in the M subunit due to mutations in the PFKM gene causes myopathy and a mild hemolytic disorder (Tarui disease or glycogen storage disease VII, OMIM 610681). PFK-deficient red blood cells display a metabolic block at the PFK step in glycolysis and have decreased concentrations of 2,3-bisphosphoglyceric acid (2,3-BPG, also called 2,3-diphosphoglyceric acid, 2,3-DPG; see later). In PFK-deficient mice, marked alterations in muscle bioenergetics and erythrocyte metabolism interact to produce a complex systemic disorder, indicating that Tarui disease is not simply a muscle glycogenosis.67 To date, 15 different mutations associated with PFK deficiency have been identified. Most are missense mutations or mutations affecting pre-mRNA processing.64,128 Approximately one third of PFK-deficient patients are of Ashkenazi Jewish origin. In this population, the most frequently encountered mutations are an intronic splice-site mutation in intron 5 and a single base pair deletion in exon 22.143,165 Most other mutations are confined to unique families. PFK is relatively unstable, and assays should be carried out on fresh blood samples. Aldolase (fructose-bisphosphate aldolase; EC 4.1.2.13) catalyzes the reversible conversion of fructose 1,6-bisphosphate to glyceraldehyde 3-phosphate and dihydroxyacetone phosphate. The enzyme is a tetramer of identical subunits of 40 kDa. Three isoenzymes have been identified to date: aldolases A, B, and C. Aldolase A is the isoenzyme that is expressed in the RBC, but also in muscle and brain. The flexible C-terminal region of aldolase A has been implicated in the catalytic function of the enzyme.53,59 Aldolase activity is markedly influenced by red cell age. The gene for aldolase A (ALDOA) is located on chromosome 16p11.2. It spans 7.5 kb and contains 12 exons. Several transcription initiation sites were identified that direct tissue-specific splicing.82 Aldolase deficiency (OMIM 611881) is a very rare disease; only six cases have been described.29,59,99,100,199 The hallmark of aldolase deficiency is chronic hemolytic anemia. In some patients, hemolysis is the sole clinical feature,99 whereas in other patients, hemolytic anemia is accompanied by myopathy,29,59,100 rhabdomyolysis,199 psychomotor retardation,100 or mental retardation.29,100 Triose-phosphate isomerase (TPI; EC 5.3.1.1) is the enzyme of the anaerobic glycolytic pathway with the highest activity. This ubiquitously expressed enzyme catalyzes the interconversion of glyceraldehyde 3-phosphate and dihydroxyacetone phosphate. TPI is active as a dimer, consisting of two identical 27 kDa subunits of 248 amino acids. The three-dimensional structure of the human enzyme has been obtained by crystallography.109 No isoenzymes are known; only three distinct electrophoretic forms attributable to minor post-translational modifications have been identified.141,200 In red blood cells, TPI activity is not related to red cell age. TPI is transcribed from a single gene (TPI1), located on chromosome 12p13. The gene spans more than 5 kb and contains 7 exons that encode the 248 amino acids–long TPI subunit.39 TPI deficiency (OMIM 190450) is a relatively rare autosomal recessive multisystem disorder, characterized by hemolytic anemia, severe neuromuscular defects, increased susceptibility to infection, and cardiomyopathy.48,134,161 Patients usually die in childhood, although intriguing exceptions have been reported.76 Because of metabolic block at the TPI step, a 20- to 60-fold increase in red blood cell concentration of dihydroxyacetone phosphate occurs.155 Lipid abnormalities have been implicated in the complex pathogenesis of TPI deficiency.77 To date, 14 mutations have been identified in the gene encoding TPI.48,133 Mutations result in decreased enzymatic activity and/or dissociation of the TPI dimer into inactive monomers.133,134 The most common mutation produces a glutamic acid-to-aspartic acid substitution of residue 104—a mutation detected in several unrelated families. Haplotype analyses suggest a single origin for this mutation with the common ancestor in Northern Europe.160 The other mutations are detected only in individual families. Mutations have been reported in TPI promoter sequences of putative regulatory importance, but they exert little if any effect on TPI enzyme activity.181 Phosphoglycerate kinase (PGK; EC 2.7.2.3) catalyzes the reversible conversion of 1,3-bisphosphoglycerate (also called 1,3-diphosphoglycerate) to 3-phosphoglycerate, thereby generating one molecule of ATP. The reaction can be bypassed by the Rapoport-Luebering shunt at the expense of one molecule of ATP (see later and Figure 23-1). This alternative routing of glycolytic intermediates has been called the energy clutch of glycolysis.96 In humans, two isoenzymes (PGK-1 and PGK-2) exist. PGK-1 is ubiquitously expressed in all somatic cells; PGK-2 is expressed only in spermatozoa.118 PGK-1 is a monomeric enzyme consisting of 417 amino acids.78 The gene encoding PGK-1 (PGK1) is located on the long arm of the X chromosome (Xq13). The gene spans 23 kb and is composed of 11 exons.122 PGK deficiency (OMIM 311800) is one of the uncommon causes of HNSHA. Sometimes the deficiency manifests only chronic hemolytic anemia, but in many cases, other clinical findings are present, particularly mental retardation and muscle disease; these may occur with or without the anemia.19 Twenty-eight families with clinical symptoms of severe PGK deficiency have been described, and in 20 of these families the causative mutation is known.19,167,170 Most of the mutations are missense ones. They are all unique except for the c.491A>T (p.Asp164Val) change, which has been encountered three times, each in the context of a different haplotype,62 suggesting independent origins of this mutation.19 One additional PGK variant (PGK München, p.Asp268Asn) does not seem to result in any clinical or biochemical abnormalities.63 Review of amino acid substitutions in PGK and the crystal structure of pig muscle PGK61 revealed that most missense mutations occur in three specific regions of the molecule. No correlation could be found, however, between these different locations and clinical expression of the enzyme deficiency.130,167 Hence, the reason for the range of manifestations in PGK deficiency remains unclear. Pyruvate kinase (PK; EC 2.7.1.40) catalyzes the conversion of phosphoenolpyruvate to pyruvate with concomitant generation of the second molecule of ATP in glycolysis (see Figure 23-1). Pyruvate is crucial for several metabolic pathways, and PK represents one of the major regulatory enzymes of glycolysis. PK is allosterically activated by its substrate and by FBP (fructose 1,6-bisphosphate), and its enzymatic activity strongly depends on red blood cell age. Therefore, because the youngest red blood cells have the highest activity, a deficiency of PK may be masked by reticulocytosis. PK is a homotetrameric enzyme. In mammals, four isozymes are expressed: PK-M1 is expressed in skeletal muscle, heart, and brain. It is the only PK isozyme that is not allosterically regulated. PK-M2 is expressed in early fetal tissues and in adult tissues including leukocytes and platelets. Both M1 and M2 subunits are produced from a single gene (PKM2) by means of alternative splicing.131,173 PK-L is predominantly expressed in the liver, whereas expression of PK-R is confined to the red blood cell.89 The PK-L and PK-R subunits are transcribed from a single gene (PKLR, for pyruvate kinase, liver, and RBC) located on chromosome 1q21, by the use of tissue-specific promoters.89,132 The PKLR gene consists of 12 exons and is approximately 9.5 kb in size.104 Exon 1 is exclusively expressed in erythroid cells, whereas expression of exon 2 is confined to the liver. Hence, the PK-R monomer is composed of 574 amino acids.88 The PK-L subunit comprises 531 amino acids.174 In basophilic erythroblasts, both PK-M2 and PK-R isozymes are expressed. During further erythroid differentiation and maturation, the PK-R isozyme progressively replaces PK-M2.116,117,172 In addition, in the red blood cell, limited proteolytic degradation of the 63 kDa PK-R subunit produces a subset of PK-R monomers of 57 to 58 kDa.86 Consequently, in young and mature human red blood cells, two distinct species can be distinguished—PK-R1 and PK-R2—that differ in PK-R and “processed” PK-R subunit composition.187 The crystal structure of human red cell PK has been elucidated.184 Each PK-R subunit is composed of four domains: N, A, B, and C. The active site lies in a cleft between the A domain and the flexible B domain. The B domain is capable of rotating with respect to the A domain, generating the “open” or “closed” conformation. The C domain contains the binding site for FBP. In the PK tetramer, subunit interactions at the interfaces between A and C domains, as well as A/B and A/C domain interactions within one subunit, are considered to be key determinants of the allosteric response, involving switching from the low-affinity T-state to the high affinity R-state.85,114,183,184,196 Pyruvate kinase deficiency (OMIM 266200) is the most common cause of nonspherocytic hemolytic anemia caused by defective glycolysis. It is an autosomal recessive disease. In the general white population, the allelic frequency is estimated to be around 2%.25 The prevalence appears to be higher among blacks.123 Two major metabolic abnormalities resulting from PK deficiency are ATP depletion and increased levels of 2,3-BPG. The precise mechanisms by which enzyme deficiency leads to a shortened RBC life span are unknown. An important feature, however, involves the selective sequestration of PK-deficient reticulocytes by the spleen.119 In addition, metabolic disturbances may affect not only red blood cell survival but also the maturation of PK-deficient erythroid progenitors, resulting in ineffective erythropoiesis.3,4 PK-deficient patients display a highly variable degree of chronic hemolysis with variable clinical severity. Clinical symptoms range from severe anemia and death at birth, severe transfusion-dependent chronic hemolysis, or moderate hemolysis with exacerbation during infection, to a well-compensated hemolysis without anemia.204 Splenectomy is, in general, beneficial.204 PK deficiency has been treated successfully by stem cell transplantation.175 Gene therapy strategies have been shown to be able to correct the PK-deficient phenotype in mice.92,121 To date, more than 190 mutations in PKLR have been reported to be associated with pyruvate kinase deficiency (www.pklrmutationdatabase.com). Most (70%) of these mutations are missense mutations affecting conserved residues in structurally and functionally important domains of PK. The most frequently detected mutations are missense mutants c.1456C>T (p.Arg486Trp), c.1529G>A (p.Arg510Gln), and c.994G>A (p.Gly332Ser), and nonsense mutant c.721G>T (p.Glu241X). Evaluating the protein structural context of affected residues using the three-dimensional structure of recombinant human tetrameric PK has provided a rationale for the observed enzyme deficiency and contributes to a better understanding of the genotype-to-phenotype correlation in PK deficiency.184,185,203 It is important to note that because most PK-deficient patients are compound heterozygous for two different (missense) mutations, up to seven different tetrameric forms of PK may be present in such patients, each with distinct structural and kinetic properties. This complicates genotype-to-phenotype correlations, as it is difficult to infer which mutation is primarily responsible for deficient enzyme function and the clinical phenotype.185 Pyruvate kinase deficiency has a protective effect against replication of the malarial parasite in human erythrocytes.13,58 This protective effect may be related to reduced ATP levels in PK-deficient red blood cells.12 Normally, approximately 10% of glucose is catabolized through the hexose monophosphate pathway (see Figure 23-1). The primary function of this pathway is to reduce 2 moles of NADP+ to NADPH, by means of oxidizing G6P. The amount of glucose passing through this pathway is regulated by the amount of NADP+ that has been made available by the oxidation of NADPH. In the red blood cell, NADPH is required mainly for the regeneration and preservation of the reduced form of glutathione (GSH), which is crucial to the cell in detoxifying hydrogen peroxide and, thereby, protecting against oxidative stress. NADPH also binds to catalase, thereby affecting its activity.163 Because the red blood cell has no other way of generating NADPH, it depends strongly on the activity of the prime enzyme of NADPH production: glucose-6-phosphate dehydrogenase (G6PD). The active enzyme is predominantly a homodimer that comprises 59 kDa subunits of 515 amino acids each. Lowering the pH causes a shift from the dimeric to the tetrameric form.49 In the absence of NADP+, G6PD dissociates into inactive subunits. Each G6PD subunit is built up by two domains. The extensive interface between the two monomers is of crucial importance for enzymatic stability and activity.11 The importance of NADP+ for stability is explained by the structural NADP+ site, distant from the active site but close to the dimer interface.11 The gene coding for G6PD is located on the long arm of the X chromosome (Xq28). It spans 18 kb and consists of 13 exons. Exon 1 is noncoding. The promoter shares many features common to other housekeeping genes.112 G6PD deficiency (OMIM 305900) is the most common enzymopathy, affecting an estimated 400 million people worldwide. The parallel between the high frequency of G6PD deficiency and the worldwide distribution of malaria implies that G6PD deficiency confers a selective advantage.177 This hypothesis is supported by several lines of evidence,40,108 indicating that the uniform state of G6PD deficiency in hemizygous males, and possibly homozygous females, confers significant protection against severe, life-threatening malaria.73,157 G6PD variants have been grouped into five categories according to the level of enzyme activity and clinical manifestations (Table 23-1).1 More than 400 variants and 140 different mutations have been reported, most of which encode the substitution of a single amino acid. The most common deficient variant is G6PD A−. This variant is characterized by an Asn-to-Asp substitution at codon 126 (c.376A>G), in most cases accompanied by a second mutation encoding a Val-to-Met change at codon 68 (c.202G>A). The second most common variant is G6PD Mediterranean, characterized by a Ser-to-Phe substitution at codon 188 (c.563C>T). Two other polymorphic variants are G6PD Seattle (c.844G>C; p.Asp282His) and G6PD Union (c.1360C>T; p.Arg454Cys). TABLE 23-1 Classes of Severity of Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency1 Most G6PD-deficient individuals are asymptomatic throughout life. They develop acute hemolysis only during periods of increased oxidative stress, elicited by certain drugs,18 infection, or the ingestion of fava beans. Clinically, such hemolytic episodes are characterized by anemia and jaundice accompanied by increased concentrations of bilirubin and LDH in plasma and reticuloytosis. The exact mechanism by which increased oxidative stress leads to acute hemolysis is unknown, but it results from the inability of G6PD-deficient red blood cells to withstand oxidative damage induced by the triggers mentioned previously. In general, older red blood cells are more susceptible to destruction than young ones. Consequently, they are selectively removed from the circulation, characterizing a self-limited course of hemolysis.57 The reticulocytosis that is elicited as a result of hemolysis may obscure the enzyme deficiency, as these very young red blood cells have the highest enzyme activity. Notably, as a result of (skewed) X chromosome inactivation, heterozygous females may be susceptible to the same pathophysiologic phenotype as their hemizygous male counterparts.30,93,156 G6PD-deficient neonates who have coinherited a mutation in the uridine-diphosphate-glucuronosyl-transferase-1 (UGT1A1) promoter are particularly at risk for developing neonatal jaundice and even kernicterus.94,205 A small proportion of G6PD-deficient individuals display the phenotype of chronic nonspherocytic hemolytic anemia. The hemolytic anemia in these class I G6PD-deficient patients may be severe. Mutations associated with class I G6PD deficiency are clustered in exons 10 and 11, designating the subunit interface.11 For further reading on the extensive subject of G6PD deficiency, we refer the reader to excellent reviews.17,41,107,189 Even though the oxidation of phosphogluconate to ribulose 5-phosphate by 6-phosphogluconate dehydrogenase generates one mole of NADPH, the few cases of 6-phosphogluconate dehydrogenase deficiency usually are not associated with hemolysis.42 Red blood cell 2,3-BPG (also called 2,3-DPG) is important in the regulation of the oxygen affinity of hemoglobin.15,16,180 2,3-BPG is synthesized and dephosphorylated in the Rapoport-Luebering shunt. Thus, this unique glycolytic bypass represents an important physiologic means for the regulation of oxygen affinity. At the same time, the Rapoport-Luebering shunt provides the red cell with flexibility with regard to the generation of ATP. From an oxygen transport point of view, the Embden-Meyerhof pathway serves principally the generation of 2,3-BPG, because in terms of quantity, it is the principal glycolytic intermediate: the concentration of 2,3-BPG is about equal to the sum of all other glycolytic intermediates. In PK deficiency, 2,3-BPG is increased as a result of the metabolic block at the PK step and of retrograde accumulation of products of glycolysis. Increased 2,3-BPG levels result in decreased oxygen affinity for hemoglobin, so that oxygen is more readily transferred to tissue. This beneficial circumstance is absent in those glycolytic enzyme defects that cause a decrease in 2,3-BPG levels (e.g., HK and GPI deficiency). Both reactions in the Rapoport-Luebering shunt are catalyzed by one multifunctional protein: 2,3-BPG mutase (BPGM) (EC 5.4.2.4).81,153 The mutase activity of this enzyme converts the glycolytic intermediate 1,3-BPG to 2,3-BPG. BPGM also has phosphatase activity (2,3-BPG phosphatase [BPGP]; EC 3.1.3.13), converting 2,3-BPG to 3-phosphoglycerate, which then re-enters the glycolytic pathway (see Figure 23-1). In addition, 2,3-BPG may be converted to 2-phosphoglycerate by multiple inositol polyphosphate phosphatase (MIPP1; EC 3.1.3.62), an enzyme that has recently been shown to expand the regulatory capacity of the Rapoport-Luebering shunt.45 BPGM is a homodimer with 30 kDa subunits consisting of 258 amino acids. Elucidation of the crystal structure of human BPGM has provided a structural basis for the different enzymatic activities of this enzyme.190 The gene for BPGM (BPGM) is located on chromosome 7q31-34. It consists of three exons, spanning more than 22 kb.84 It is expressed only in erythroid tissue during the late stages of differentiation.84 BPGM deficiency (OMIM 222800) is a very rare autosomal recessive disorder, resulting in markedly decreased concentrations of 2,3-BPG in erythrocytes. As a result, the oxygen affinity of hemoglobin is increased, causing a decrease in tissue oxygenation and, consequently, erythrocytosis. Only a few cases of BPGM deficiency have been described, all belonging to one, clinically normal French family.154 Two mutations have been identified in BPGM. One single nucleotide substitution predicts the substitution of the highly conserved Arg89 by Cys (BPGM Creteil I).154 BPGM Creteil II is characterized by a deletion of nucleotide 205, resulting in a shift in the reading frame and, consequently, leading to a premature stop codon.103
Enzymes of the Red Blood Cell
The Embden-Meyerhof Pathway
Hexokinase
Glucose-6-Phosphate Isomerase
Phosphofructokinase
Aldolase
Triose-Phosphate Isomerase
Phosphoglycerate Kinase
Pyruvate Kinase
Hexose Monophosphate Pathway
Glucose-6-Phosphate Dehydrogenase
Class
Description
Class I
Severe deficiency associated with chronic hemolytic anemia
Class II
Severe deficiency (<10% residual activity), usually without hemolytic anemia
Class III
Moderate to mild deficiency (10 to 60% residual activity) (e.g., G6PD A)
Class IV
Very mild or no deficiency (e.g., G6PD A)
Class V
Increased activity (only one such variant has been described—G6PD Hektoen)
6-Phosphogluconate Dehydrogenase (Decarboxylating) (EC 1.1.1.44)
Rapoport-Luebering Shunt
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


