Environmental Toxicology: Carcinogens and Heavy Metals
Myriad authoritative textbooks are available in the area of environmental toxicology. This chapter does not attempt a thorough coverage; rather, it sets forth a few basic principles, briefly discusses carcinogens and chemoprevention, and then focuses on the pharmacotherapy of heavy metal intoxication.
ASSESSMENT AND MANAGEMENT OF ENVIRONMENTAL RISK
When assessing the risks of environmental exposures to xenobiotics, one must consider population exposures to low-dose toxicants over long periods of time. Thus, one must give careful attention to the low end of the dose-response curve, using experiments based on chronic exposures. Unlike drugs, which are given to treat a specific disease and should have benefits that outweigh the risks, environmental toxicants usually are only harmful. In addition, exposures to environmental toxicants usually are involuntary, there is uncertainty about the severity of their effects, and people are much less willing to accept their associated risks.
Epidemiology and toxicology are 2 approaches used to predict the toxic effects of environmental exposures. Epidemiologists monitor health effects in humans and use statistics to associate those effects with exposure to an environmental stress, such as a toxicant. Toxicologists perform laboratory studies to try to understand the potential toxic mechanisms of a chemical to predict whether it is likely to be toxic to humans. Information from both approaches is integrated into environmental risk assessment. Risk assessment is used to develop laws and regulations to limit exposures to environmental toxicants to a level that is considered safe.
EPIDEMIOLOGICAL APPROACHES TO RISK ASSESSMENT
Assessing human exposures over long periods of time and drawing conclusions about the health effects of a single toxicant present great challenges. Epidemiologists usefully rely on biomarkers in assessing risk. There are 3 types of biomarkers:
• Biomarkers of exposure usually are measurements of toxicants or their metabolites in blood, urine, or hair. Blood and urine concentrations measure recent exposures, while hair levels measure exposure over a period of months. An example of an unusual exposure biomarker is X-ray fluorescent measurement of bone lead levels, which estimates lifetime exposure to lead.
• Biomarkers of toxicity are used to measure toxic effects at a subclinical level and include measurement of liver enzymes in the serum, changes in the quantity or contents of urine, and performance on specialized exams for neurological or cognitive function.
• Biomarkers of susceptibility are used to predict which individuals are likely to develop toxicity in response to a given chemical. Examples include single nucleotide polymorphisms in genes for metabolizing enzymes involved in the activation or detoxification of a toxicant. Some biomarkers simultaneously provide information on exposure, toxicity, and susceptibility. For example, the measurement in the urine of N7-guanine adducts from aflatoxin B1 provides evidence of both exposure and a toxic effect (in this case, DNA damage). Such biomarkers are valuable because they can support a proposed mechanism of toxicity.
Several types of epidemiological studies are used to assess risks, each with its own set of strengths and weaknesses. Ecological studies correlate frequencies of exposures and health outcomes between different geographical regions. These studies can detect rare outcomes but are prone to confounding variables, including population migration. Cross-sectional studies examine the prevalence of exposures and outcomes at a single point in time. Such studies determine an association but do not provide a temporal relationship and are not effective for establishing causality. Case-control studies start with a group of individuals affected by a disease, which then is matched to another group of unaffected individuals for known confounding variables. Questionnaires often are used to evaluate past exposures. This method also is good for examining rare outcomes because the endpoint is known. However, case-control studies rely on assessments of past exposures that often are unreliable and can be subject to bias. Prospective cohort studies measure exposures in a large group of people and follow that group for a long time to measure health outcomes. These studies are good at establishing causality, but they are extremely expensive, particularly when measuring very rare outcomes, because a large study population is required to observe sufficient disease to obtain statistical significance. One of the key types of human studies used in drug development is the randomized clinical trial (see Chapter 1). Such studies cannot be used to directly measure the effects of environmental toxicants (for obvious ethical reasons) but can be used to examine the effectiveness of an interventional strategy for reducing both exposure to toxicants and disease.
TOXICOLOGICAL APPROACHES TO RISK ASSESSMENT
Toxicologists use model systems, including experimental animals, to examine the toxicity of chemicals and predict their effect on humans. The significance of these model systems to human health is not always established. Toxicologists also test chemicals at the high end of the dose-response curve in order to see enough occurrences of an outcome to obtain statistical significance. As a result, there often is uncertainty about the effects of very low doses of chemicals. To determine the applicability of model studies, toxicologists study the mechanisms involved in the toxic effects of chemicals.
To predict the toxic effects of environmental chemicals, toxicologists perform subchronic (3 months of treatment for rodents) and chronic studies (2 years for rodents) in at least 2 different animal models. Subchronic experiments provide a model for occupational exposures, while chronic experiments are used to predict effects from lifetime exposures to chemicals in food or the environment. Doses are chosen with the goal of having 1 concentration that does not have a significant effect, 1 concentration that results in statistically significant toxicity at the low end of the dose-response curve, and 1 or more concentrations that will have moderate-to-high levels of toxicity. A theoretical dose-response curve for an animal study is shown in Figure 67–1. An animal study provides 2 numbers that estimate the risk from a chemical. The no observed adverse effect level (NOAEL) is the highest dose used that does not result in a statistically significant increase in negative health outcomes. The lowest observed adverse effect level (LOAEL) is the lowest dose that results in a significant increase in toxicity. The NOAEL is divided by 10 for each source of uncertainty to determine a reference dose (RfD), which is commonly used as a starting point for determining regulations on human exposures to chemicals. The modifiers used to determine the RfD are based on the uncertainties between the experimental and human exposure. The most common modifiers used are for interspecies variability (human to animal) and interindividual variability (human to human), in which case RfD = NOAEL/100. When a NOAEL is unavailable, a LOAEL may be used, in which case another 10-fold uncertainty factor is used. The use of factors of 10 in the denominator for determination of RfD is an application of the “precautionary principle,” which attempts to limit human exposure by assuming a worst-case scenario for each unknown variable. Animal studies typically are designed to obtain statistical significance with a 10% increase in an outcome. As a result, there is considerable uncertainty about what occurs below that level, as demonstrated in Figure 67–1. Toxicologists often assume that there is a threshold dose (T), below which there is no toxicity. However, many carcinogens and other toxicants with specific molecular targets (e.g., lead) do not exhibit a threshold. Ideally, mechanistic studies should be done to predict which dose-response curve is most likely to fit a given chemical.
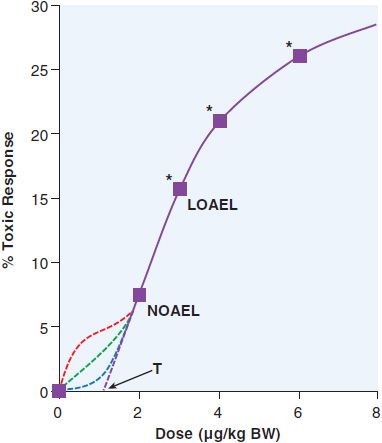
Figure 67–1 LOAEL and NOAEL. The theoretical dose-response curve from an animal study demonstrates the no observed adverse effect level (NOAEL) and the lowest observed adverse effect level (LOAEL). Below the NOAEL level, there is considerable uncertainty as to the shape of the response curve. It could continue linearly to reach a threshold dose (T) where there would be no harmful effects from the toxicant, or it could have a number of different possible inflection points. Each of these curves would reflect very different effects on human populations. *, statistically significant; BW, body weight.
Toxicologists perform a variety of mechanistic studies to understand how a chemical might cause toxicity. Computer modeling using a compound’s 3-dimensional structure to determine quantitative structure-activity relationships (QSARs) is commonly performed on both drugs and environmental chemicals. QSAR approaches can determine which chemicals are likely to exhibit toxicities or bind to specific molecular targets. Cell-based approaches in prokaryotes and eukaryotes are used to determine whether a compound damages DNA or causes cytotoxicity. DNA damage and the resulting mutagenesis often are determined with the Ames test. The Ames test uses Salmonella typhimurium strains with specific mutations in the gene needed to synthesize histidine. These strains are treated with chemicals in the presence or absence of a metabolic activating system, usually the supernatant fraction from homogenized rat liver. If a compound is a mutagen in the Ames test, it reverts the mutation in the histidine operon and allows the bacteria to form colonies on plates with limited histidine. Gene chip microarrays assess gene expression in cells or tissues from animals treated with a toxicant and provide a very useful tool to identify the molecular targets and pathways altered by toxicant exposures. The susceptibility of knockout mice to a toxicant can help to determine whether the knocked-out genes are involved in the metabolic activation and detoxification of a given toxicant.
CARCINOGENS AND CHEMOPREVENTION
CARCINOGENESIS
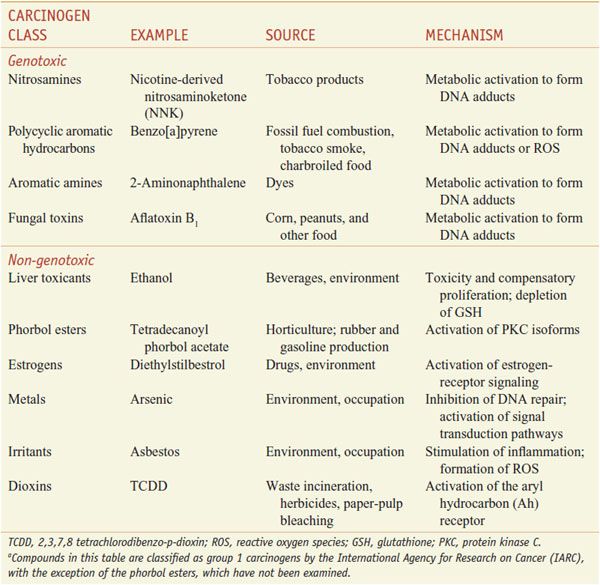
The International Agency for Research on Cancer (IARC) classifies compounds into groups based on risk assessments using human, animal, and mechanism data. Chemicals in group 1 are known human carcinogens; group 2A includes chemicals that probably are carcinogenic in humans; group 2B chemicals possibly are carcinogenic in humans; group 3 chemicals lack data to suggest a role in carcinogenesis; and group 4 are those with data indicating they are unlikely to be carcinogens. Table 67–1 presents information of some Group 1 carcinogens.
Table 67–1
Examples of Important Carcinogensa
The transformation of a normal cell to a malignancy is a multi-stage process, and exogenous chemicals can act at 1 or more of these stages (Figure 67–2). A classic model of chemical carcinogenesis is tumor initiation followed by tumor promotion. A tumor initiator causes gene mutations that increase the ability of cells to proliferate and avoid apoptosis. A tumor promoter does not directly modify genes but changes signaling pathways and/or the extracellular environment to cause initiated cells to proliferate. Although this model is an oversimplification, it demonstrates the types of changes that must occur to transit a normal cell into tumorigenesis.
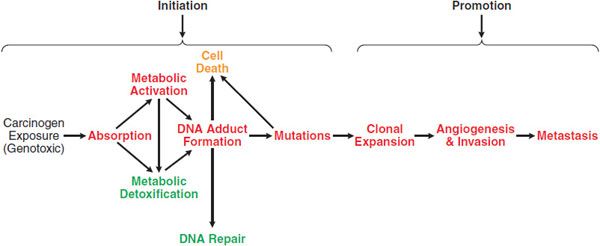
Figure 67–2 Carcinogenesis: initiation and promotion. There are many steps that occur between the exposure to a genotoxic carcinogen and the development of cancer. Processes in red lead to the development of cancer, while those in green reduce the risk. Non-genotoxic carcinogens act by enhancing steps leading to cancer and/or inhibiting protective processes. A chemopreventive agent acts by inhibiting steps leading to cancer or by increasing protective processes.
Chemical carcinogens cause cancer through genotoxic and non-genotoxic mechanisms (Figure 67–2). Genotoxic carcinogens induce tumor formation through damage to DNA. Typically, genotoxic carcinogens undergo metabolism in a target tissue to a reactive intermediate. This reactive intermediate can directly damage DNA via covalent reaction to form a DNA adduct. Alternatively, it can indirectly damage DNA through the formation of reactive oxygen species (ROS), which can oxidize DNA or form lipid peroxidation products that react with DNA. If DNA damage from a genotoxic carcinogen is not repaired prior to DNA replication, a mutation can result. If this mutation is in a key tumor suppressor gene or proto-oncogene, it provides advantages in proliferation or survival. Alternatively, if the mutation is in a DNA repair gene, the mutation increases the probability that other mutations will occur. Genotoxic carcinogens are tumor initiators.
Benzo[a]pyrene, a key carcinogen in tobacco smoke, is an example of a genotoxic carcinogen that forms both direct DNA adducts and ROS. Benzo[a]pyrene is oxidized by CYPs to a 7,8-dihydrodiol, which represents a proximate carcinogen (a more carcinogenic metabolite). This metabolite can either undergo a second oxidation step by a CYP to form a diol epoxide, which readily reacts with DNA, or it can undergo oxidation by aldo-keto reductases to form a catechol, which will redox cycle to form ROS.
Non-genotoxic carcinogens increase the incidence of cancer without damaging DNA. Many non-genotoxic carcinogens bind to receptors that stimulate proliferation or other tumor-promoting effects, such as tissue invasion or angiogenesis. For example, phorbol esters mimic diacylglycerol and activate PKC isoforms. This in turn stimulates MAP kinase pathways, leading to proliferation, invasiveness, and angiogenesis (see Chapter 3). In most normal cells, prolonged activation of this pathway stimulates apoptosis, but cells with defective apoptotic mechanisms due to preceding mutation(s) are resistant to this effect. Estrogenic carcinogens activate estrogen receptor-α (ERα) and stimulate proliferation and invasiveness of estrogen responsive cells. Chronic inflammation is another mechanism of non-genotoxic carcinogenesis. Inflammatory cytokines stimulate PKC signaling, leading to proliferation, invasiveness, and angiogenesis. Irritants such as asbestos are examples of carcinogens that work through inflammation. Chronic exposure to hepatotoxic chemicals (or chronic liver diseases) also causes non-genotoxic carcinogenesis by stimulating compensatory proliferation to repair the liver damage. This damage and repair process increases the likelihood of DNA damage becoming a mutation, causes chronic inflammation, and selects for cells that proliferate faster or are less sensitive to apoptosis.
Tumor initiation also may occur through non-genotoxic mechanisms. For example, some heavy metals do not directly react with DNA but interfere with proteins involved in DNA synthesis and repair, increasing the likelihood that an error will be made during replication. Non-genotoxic carcinogens also can cause heritable changes to gene expression by altering the methylation state of cytosines in 5’-CpG-3 islands of gene promoters, thus acting as tumor initiators. Methylation can silence tumor suppressor genes, while demethylation of protooncogenes can increase their expression.
CHEMOPREVENTION
Drugs that interfere with the carcinogenic process to prevent cancer before it is diagnosed are termed chemopreventive agents. Chemoprevention strategies often are based on epidemiological studies of nutrition, where there are many examples of clear protective effects of plant-based foods and drinks on the incidence of various types of cancer. A number of compounds for the prevention of cancer are in clinical trials (Table 67–2). There currently are no drugs approved for chemoprevention of environmental carcinogenesis, but there are approved drugs to prevent carcinogenesis due to endogenous estrogen (tamoxifen and raloxifene) and viruses (hepatitis B and human papillomavirus vaccines).
Table 67–2
Chemopreventive Agents Being Studied in Humans
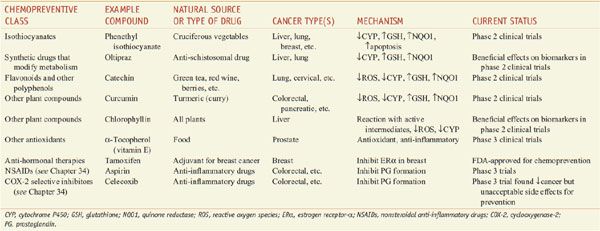
Chemopreventive agents interfere with the processes of initiation and promotion (see Figure 67–2). One mechanism of anti-initiation is prevention of carcinogen activation. Isothiocyanates and similar compounds inhibit CYPs involved in activating many carcinogens and also upregulate genes controlled by the antioxidant response element (ARE); the ARE-responsive group includes γ-glutamylcysteine synthase light chain (the gene responsible for the rate-determining step in GSH synthesis) and quinone reductase (NQO1). Increased expression of ARE-regulated genes is predicted to increase the detoxification of proximate carcinogens. Isothiocyanates also stimulate apoptosis of p53-deficient cells via the formation of cytotoxic DNA adducts. Compounds that act as antioxidants may provide protection because many carcinogens work through the generation of ROS. Some compounds simultaneously prevent carcinogen activation and act as antioxidants. For example, flavonoids and other polyphenols found in a wide variety of plants are potent antioxidants that inhibit CYPs and induce expression of ARE-regulated genes. Chlorophyll and other compounds can protect against carcinogens by binding to or reacting with carcinogens or their metabolites and prevent them from reaching their molecular target.
Inflammation is a potential target for chemoprevention through interference with promotion. The COX-2 inhibitor celecoxib has demonstrated efficacy at reducing the risk of colorectal cancer. However, this benefit was offset by an increased risk of death due to cardiovascular events, forcing the early termination of the trial. Studies examining long-term treatment with aspirin for cardiovascular benefits found that aspirin also reduces the incidence of colorectal adenomas. Natural compounds, such as α-tocopherol, also can exert chemoprevention by reducing inflammation.
One successful approach to chemoprevention is modification of nuclear receptor signaling. Promising preliminary data suggested that retinoids might be beneficial for preventing lung and other cancers. The selective ER modulators tamoxifen and raloxifene reduce the incidence of breast cancer in high-risk women and are approved for chemoprevention in these patients.
AFLATOXIN B1. Promising agents are being developed as chemopreventants of hepatocarcinogenesis mediated by aflatoxin B1. Aflatoxins are produced by Aspergillus flavus, a fungus that is a common contaminant of foods, especially corn, peanuts, cottonseed, and tree nuts. A. flavus is abundant in regions with hot and wet climates.
ADME. Aflatoxin B1 is readily absorbed from the GI tract and initially distributed to the liver, where it undergoes extensive first-pass metabolism. Aflatoxin B1 is metabolized by CYPs, including 1A2 and 3A4, to yield either an 8,9-epoxide or products hydroxylated at the 9 position (aflatoxin M1) or 3 position (aflatoxin Q1; Figure 67–3). The hydroxylation products are less susceptible to epoxidation and are therefore detoxification products. The 8,9-epoxide is highly reactive toward DNA and is the reactive intermediate responsible for aflatoxin carcinogenesis. The 8,9-epoxide is short lived and undergoes detoxification via nonenzymatic hydrolysis or conjugation with GSH. Aflatoxin M1 enters the circulation and is excreted in urine and milk. Hydroxylated aflatoxin metabolites also can undergo several additional phase 1 and phase 2 metabolic pathways prior to excretion in urine or bile.
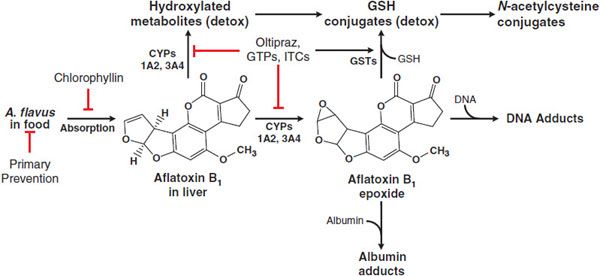
Figure 67–3 Metabolism and actions of aflatoxin B1. Following ingestion and absorption of food containing A. flavus, aflatoxin B1 undergoes activation by CYPs to its 8,9-epoxide, which can be detoxified by glutathione S-transferases (GSTs) or by spontaneous hydration. Alternatively, it can react with cellular macromolecules such as DNA and protein, leading to toxicity and cancer. Oltipraz, green tea polyphenols (GTPs), and isothiocyanates (ITCs) decrease aflatoxin carcinogenesis by inhibiting the CYPs involved aflatoxin activation and increasing the synthesis of GSH for GSTs involved in detoxification.
Toxicity. Aflatoxin B1 primarily targets the liver, although it also is toxic to the GI tract and hematological system. High-dose exposures result in acute necrosis of the liver, leading to jaundice and, in many cases, death. Acute toxicity from aflatoxin is relatively rare in humans and requires consumption of milligram quantities of aflatoxin per day for multiple weeks. Chronic exposure to aflatoxins results in cirrhosis of the liver and immunosuppression.
Carcinogenicity. Based on increased incidence of hepatocellular carcinoma in humans exposed to aflatoxin and supporting animal data, IARC has classified aflatoxin B1 and several other natural aflatoxins as known human carcinogens (group 1). Aflatoxin exposure and hepatitis B virus work synergistically to cause hepatocellular carcinoma. Separately, aflatoxin or hepatitis B exposure increases the risk of hepatocellular carcinoma 3.4- or 7.3-fold, respectively; those exposed to both have a 59-fold increased risk of cancer compared to unexposed individuals. Aflatoxin primarily forms DNA adducts at deoxyguanosine residues, reacting at either the N1 or N7 position. The N7-guanine adduct mispairs with adenine, leading to G → T transversions. Human aflatoxin exposure is associated with hepatocellular carcinomas bearing an AGG to AGT mutation in codon 249 of the p53 tumor suppressor gene, resulting in the replacement of an arginine with cysteine.
The interaction between aflatoxin and hepatitis B that is responsible for the increased incidence of hepatocellular carcinoma is not well understood. Hepatitis B influences the metabolism of aflatoxin B1 by upregulating CYPs, including 3A4, and decreasing glutathione S-transferase activity. In addition, hepatocellular proliferation to repair damage done by hepatitis B infection increases the likelihood that aflatoxin-induced DNA adducts will cause mutations. The hepatotoxic and tumor-promoting effects of hepatitis B also could provide a more favorable environment for the proliferation and invasion of initiated cells.
Chemoprevention of Aflatoxin-Induced Hepatocellular Carcinoma. Inhibiting CYP activity or increasing glutathione conjugation will reduce the intracellular concentration of the 8,9-epoxide and thus prevent DNA adduct formation. One drug that has been tested as a modifier of aflatoxin metabolism is oltipraz. Oltipraz, an anti-schistosomal drug, potently inhibits CYPs and induces genes regulated by the ARE. Oltipraz increases the excretion of aflatoxin N-acetylcysteine, indicating enhanced glutathione conjugation of the epoxide. At 500 mg/wk, oltipraz reduced the levels of aflatoxin M1, consistent with inhibition of CYP activity.
Green tea polyphenols also have been used to modify aflatoxin metabolism in exposed human populations. Individuals receiving a daily dose of 500 or 1000 mg (equivalent to 1 or 2 L of green tea) demonstrated a small decline in the formation of aflatoxin–albumin adducts and a large increase in the excretion of aflatoxin N-acetylcysteine, consistent with a protective effect.
Another approach used for the chemoprevention of aflatoxin hepatocarcinogenesis is the use of “interceptor molecules.” Chlorophyllin, an over-the-counter mixture of water-soluble chlorophyll salts, binds tightly to aflatoxin in the GI tract, forming a complex that is not absorbed. In vitro, chlorophyllin inhibits CYP activity and acts as an antioxidant. In a phase 2 trial, administration of 100 mg of chlorophyllin with each meal reduced aflatoxin–N7-guanine adduct levels in the urine by >50%. Because of the strong interaction between hepatitis B and aflatoxin in carcinogenesis, the hepatitis B vaccine will reduce the sensitivity of people to the induction of cancer by aflatoxin. Primary prevention of aflatoxin exposure through hand or fluorescent sorting of crops to remove those with fungal contamination can also reduce human exposure. A more cost-effective primary prevention approach is to improve food storage to limit the spread of A. flavus, which requires a warm and humid environment.
METALS
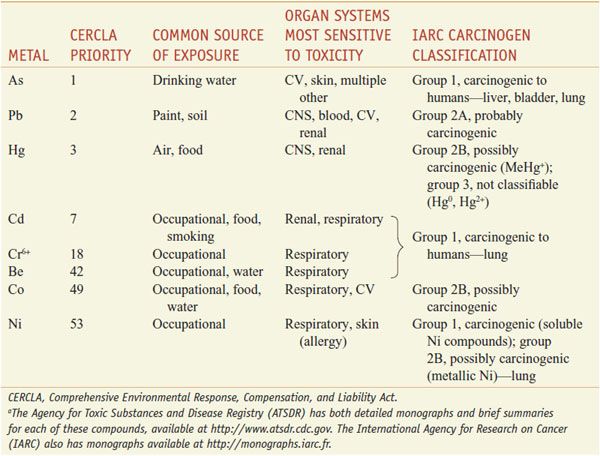
Arsenic, lead, and mercury are the top 3 substances of concern due to their toxicity and likelihood of human exposure as listed under the Comprehensive Environmental Response, Compensation, and Liability Act (CERCLA, also known as Superfund).
Many of the toxic metals in the environment also are carcinogens (Table 67–3). In addition, several essential metals also are toxic under conditions of overdose. Copper and especially iron are associated with toxicities, primarily targeting the liver through generation of ROS.
Table 67–3
Toxic Metals with Frequent Environmental or Occupational Exposurea
Not listed in Table 67–3 is the metal gold, which has its own uses and toxicities. Among heavy metals, perhaps only gold is addictive: gold has been used for centuries for relief of the itching palm, and many cannot get enough of its influence.
LEAD. Chronic exposure of populations to even very low levels of lead (Pb) has major deleterious effects that are only now beginning to be understood.
Exposure.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


