Fig. 3.1
Problems of TCRαβ gene therapy and their solution. a Formation of mispaired TCRαβ. Mispairing between endogenous TCRαβ and introduced TCRαβ may generate specificity for un-intended targets. Shown are the possible combinations generating four different TCRs that may compete CD3 complex which is needed for TCRαβ to be expressed on the T cell surface. b Reported solution for mispaired TCRαβ. Modifications to the constant region may improve desired heterodimer formation between introduced TCR chains. These include (1) use mouse constant region, (2) amino acid substitutions in constant region, and (3) insertion of another disulfide bond in constant region. Another solution uses shRNA or ZFN targeting constant region to down-regulate of prevent TCRαβ expression
Several solutions for this issue have been developed (Fig. 3.1b). As an alternative to using αβT cell, Heemskerk et al. employ γδT cells as the recipient cell for gene transfer since introduced TCRαβ chains cannot pair with endogenous TCRγδ chains [50]. However, TCRαβ-transduced TCRγδ cells typically need CD8 transduction to induce robust antigen-specific activation which may not be available on γδT cells that fail to express (CD4 and) CD8. One strategy to prevent mismatched TCRαβ formation is to facilitate heterodimer formation between the introduced TCRαβ chains. This has been accomplished (1) using full or partial mouse TCR α and β sequences [51–53], (2) introducing another disulfide bond within the introduced TCR constant regions [54, 55], and (3) designing a knob-into-hole configuration [56]. These systems have been shown to successfully reduce mismatched TCRαβ formation. However, introduced TCRαβ cannot avoid competing for the CD3 complex with endogenous TCRαβ. As a result, the copy number of introduced TCRαβ on the surface may be decreased leading to insufficient TCRαβ-mediated effector functioning.
Recently, two groups showed that endogenous TCRαβ expression can be disrupted by different molecular technologies. One team used short hairpin RNAs (shRNAs) against TCRαβ constant regions to suppress endogenous TCRαβ expression [57]. They designed a plasmid containing both shRNAs and introduced TCRαβ genes in a single retrovirus vector and demonstrated both enhanced expression of introduced TCRαβ and suppression of endogenous TCRαβ. This resulted in enhanced function of the introduced TCRαβ compared with expressing transgenic TCRαβ in T cells without shRNAs. The other team used zinc-finger nucleases (ZFNs) to completely disrupt endogenous TCRαβ expression at the genomic level [58]. ZFN employs an engineered nuclease which consists of the zinc-finger-binding domain and the cleavage domain from type II endonuclease FokI. A pair of ZFNs can induce a DNA double-strand break (DSB) at an intended genomic target site. The nonhomologous end-joining DNA DSB repair mechanism creates an indel at ZFN target sites to disrupt target gene expression by frame shift mutation. The sequential introduction of ZFNs targeting TCRα chain and then β chain was used by the authors to demonstrate that endogenous TCRαβnull WT1-specific TCRαβ+ T cells could be generated. This strategy enhances TCRαβ+ T cell function and avoids mispaired TCRαβ mediated xenogeneic GVHD in a mouse model.
3.2.1.4 TCRαβ Specificity Recapitulated Through the Introduction of a Single Chain
An alternative strategy to define TCRαβ specificity uses a single-chain TCRαβ (scTCR). This is an artificial TCRαβ chain is comprised of a variable domain, including the CDR3 region of TCRα and β chains, linked through a peptide and fused in frame to the CD3ζ signaling domain [59–61]. This technology avoids the potential for mispairing between the introduced immunoreceptor and the endogenous TCRαβ; however, the expression and affinity of scTCRαβ may be less than that of the original TCRαβ heterodimer [62]. Efforts to enhance the avidity of scTCR chain have been undertaken, and in some cases, the resultant scTCR has been shown to induce a comparable response to heterodimeric TCRαβ [56, 60, 61]. Modification of the signaling domain and increasing the affinity of the scTCR domain will needed to improve the therapeutic potential of this approach. Recently, several groups have isolated monoclonal antibodies (mAbs) having TCRαβ-like specificity [63–69]. Although the cross-reactivity of these antibodies has not been extensively studied (such as to the superstructure of HLA molecules), these mAbs may be used to construct CARs to target a peptide/HLA complex, instead of using TCRαβ gene transfer.
3.2.1.5 Clinical Trial in TCRαβ Gene Therapy
As of February 2013, five reports have been published by the NCI group describing the translation of TCR gene therapy into the clinical setting. In the seminal paper in 2006 [70], 15 patients with metastatic melanoma were treated with MART1-specific TCRαβ gene transduced autologous peripheral blood lymphocytes. These patients were treated with lymphocyte depleting chemotherapy before T cell infusions, followed by IL-2 administration and MART-1 peptide vaccination. Two of 15 patients exhibited a partial response to this therapy. A follow-up study employing the same TCRαβ specificity reported a response rate (tumor regression) of 13 % (Four out of 31 patients). A follow-up study using a high-affinity murine TCRαβ against MART-1 and gp100 resulted in a superior response (tumor regression was observed in 30 % of patients receiving MART1-specific TCRαβ-modified T cells and 19 % of patients receiving gp100-specific TCRαβ-modified T cells). However, on-target adverse events (e.g., skin rash related to destruction of melanocyte, uveitis, and vitiligo) were observed [71]. T cells genetically modified to target NY-ESO1 have also been tested in the clinic. Five of 11 patients with metastatic melanoma showed a clinical response, and two patients showed a complete response lasting more than 1 year. Furthermore, four of 6 patients with metastatic synovial sarcoma also showed a clinical response [72]. Mouse-derived TCRαβ genes against CEA have also been tested in patients with metastatic colorectal cancer. Tumor regression occurred in one out of three treated patients, but severe colitis was the dose-limiting toxicity [73]. T cells expressing a MAGE-A3-specific mouse TCRαβ have been administered, but a high incidence of neurological toxicity was observed due to unpredicted expression of MAGE-A family gene in human brain [74].
3.2.2 CAR
One of the first CARs (“T-body”) was developed by Eshhar [75]. Upon expression, T cells can recognize cell surface TAAs independent of HLA. Thus, one CAR design has application for a tumor type common to multiple patients. Recent advances in CAR design, especially in the intracellular signaling domain, the affinity of the antigen recognition domain, and the extracellular scaffold make this a promising immunotherapy. Currently, multiple clinical trials are underway to evaluate this technology.
3.2.2.1 Structure of CAR
The structure of a prototypical CAR can be divided into three segments: (1) Single chain fragment of variable (scFv) which determines the specificity, (2) extracellular spacer, and (3) intracellular signaling domain(s) (Fig. 3.2). Other aspects of CAR design may impact the ability to recycle T cell effector functions in the tumor microenvironment such as the transmembrane domain [76], where the CAR binds the TAA [77], and the density of CAR on the T cell surface.
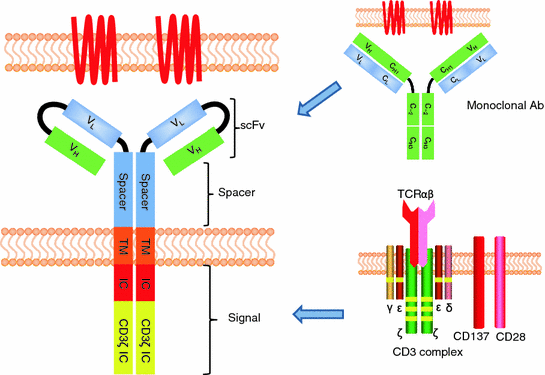
Fig. 3.2
Structure of chimeric antigen receptor. Variable regions from heavy and light chain (scFv) of mAb are linked with spacer domain and signaling domain from CD3ζ and co-stimulatory molecules (e.g., CD28 and CD137)
scFv
The scFv consists of the variable regions of heavy and light chains from a mAb with desired specificity for TAA. Indeed, this determines the specificity of the CAR. Many cell surface TAAs expressed on a variety of tumor cells have been targeted by CARs using scFvs from specific mAb (Table 3.1). In addition to these specific scFvs, a universal CAR has recently been described [78, 79]. Here, scFv from anti-FITC or avidin dimer was used to construct CARs. For example, after infusing T cells expressing a fluorescein-specific CAR, multiple cell surface molecules expressed on tumors can be targeted by infusion of FITC-labeled mAbs. This technology enables the target tumor antigens to be changed after infusing CAR-expressing T cells.
Table 3.1
Target molecules of chimeric antigen receptor for tumor therapy (as of March 2013)
Target | Target tumors | State of evaluation |
|---|---|---|
Hematological malignancies | ||
CD19 | B cell malignancies | Clinical |
CD20 | B cell malignancies | Clinical |
CD22 | B-ALL | Preclinical |
CD23 | B-CLL | Preclinical |
CD30 | Hodgkin’s lymphoma | Clinical |
CD38 | B cell malignancies including multiple myeloma | Preclinical |
CD70 | Hematological malignancies and some solid tumor (e.g. renal cell carcinoma) | Preclinical |
Igk | Mature B cell malignancies | Clinical |
ROR1 | B-CLL | Preclinical |
BCMA | Multiple myeloma | Preclinical |
CD33 | Acute myelogenous leukemia | Preclinical |
Lewis (Y) | Hematological malignancies | Clinical |
CD123 | Acute myelogenous leukemia | Preclinical |
Solid tumors | ||
EphA2 | Glioblastoma | Preclinical |
GD2 | Neuroblastoma, myeloma | Clinical |
IL11Rα | Osteosarcoma | Preclinical |
IL13Rα2 | Glioblastoma, medulloblastoma | Clinical |
EGFRvIII | Glioblastoma | Clinical |
Her2/ErbB2 | Breast cancer, osteosarcoma (low), glioblastoma, pancreatic cancer, etc | Clinical |
CAIX | Renal cell carcinoma | Clinical |
MUC1 | Hematological malignancies, many types of cancers | Preclinical |
FAP | Malignant pleural mesothelioma | Clinical |
PSMA | Prostate cancer | Clinical |
PSCA | Prostate cancer | Preclinical |
fAchR | Rhabdomyosarcoma | Preclinical |
FRα | Ovarian cancer | Clinical |
CD24 | Pancreatic cancer | Preclinical |
CEA | Colon cancer, pancreatic cancer | Clinical |
Mesothelin | Breast cancer | Clinical |
EpCAM | Many types of cancer | Preclinical |
HMW-MAA | Melanoma | Preclinical |
GUCY2C | Colon cancer | Preclinical |
GD3 | Melanoma | Preclinical |
VEGFR2 | Tumor vasculature | Clinical |
Spacer
The spacer domain is gaining importance in its ability to influence the ability of T cells to target tumor and protect the genetically modified T cells from activation-induced cell death [80]. A panel of spacers has been used including the CH2–CH3 Fc hinge domain from IgG1 or IgG4, CD8α, as well as small hinges. The IgG1 CH2–CH3 Fc hinge domain is widely used spacers, but the potential to bind to the Fcγ receptor raises concerns for deleterious side effects. Modification of this construct may avoid this issue [81]. Information regarding the impact of spacer is beginning to be published. For example, CAR-mediated activation is less efficient in T cells expressing a CAR containing a spacer sequence (IgG1 Fc domain) compared to CAR containing no spacer sequence [82]. This report was restricted to the use of first-generation (described later) signaling domain. The role that spacer sequence plays in the function of a second- or third-generation CAR constructs is being evaluated.
Intracellular domain
CD3ζ or Fc receptor γ has multiple ITAM motifs to transduce an activation signal in T cells and as such have been widely used as intracellular signaling domains. Construct containing just these ITAMs are referred to as a “first-generation” CARs. Although these signaling domains can induce an activation signal in T cells, they are usually suboptimal and curtail the therapeutic impact after adoptive transfer [83–87].
Co-stimulatory molecules that provide a “2nd signal” are required to trigger a fully competent T cell activation event. To improve CAR-mediated signaling, multiple co-stimulatory molecules have been combined with chimeric CD3ζ. CD28 is one of the well-known co-stimulatory molecules which are important for T cell survival [88]. Enforced expression of CD28 in CD28neg virus-specific CTL clones restores production of IL-2 and proliferation in these cells [89]. CD28 was first tested as a single intracellular domain instead of CD3ζ [90, 91]. T cells transduced with this construct showed increased IL-2 production and proliferation. Following this observation, Maher et al. generated prostate-specific membrane antigen (PSMA) target CAR containing both CD28 and CD3ζ signaling domain (“2nd generation”) [92]. T cells expressing this CAR construct produced increased amounts of IL-2, showed increased proliferation, and maintained specificity for target PSMA. The authors also showed that the functional activity was dependent on the order of CD28 and CD3ζ within the CAR. Improved function was observed in the CAR having transmembrane and proximal intracellular domain of CD28 fused to CD3ζ. The improved function afforded by the CAR design that employs CD28 and CD3ζ CAR has been confirmed in vitro [83, 93] and in the clinical setting [94]. Co-stimulatory molecules belonging to the tumor necrosis factor (TNF) ligand superfamily (e.g., 4-1BB (CD137), OX40 (CD134), and CD27) are also important for co-stimulation of T cells and promote long-term T cell survival [95, 96]. These molecules associate with TNF receptor-associated factor (TRAF) and link to many molecules which are important for cell survival (e.g., BCL2, BCL-X, phosphoinositide 3 kinase, AKT, and NFkB pathway) [97]. As anticipated, CAR constructs containing chimeric CD134 [98, 99] or CD137 [100–102] have been shown to sustain in vivo persistence of CAR+ T cells. Combining signaling from multiple signaling molecules, such as CD3, CD28, and CD137 (or CD134) to form a third-generation CAR, has also been tested [103–105]. Several groups have reported that CAR containing three activation motifs have potent anti-tumor efficacy in models [106, 107]. A CAR signaling through chimeric CD27 has also been shown to result in improved function and in vivo survival, comparable to CAR containing CD137 [108]. In addition to anti-apoptotic molecules, CD27 has been reported to activate the Wnt pathway in chronic myeloid leukemia stem cells [109]. Since the Wnt pathway has recently been identified as the key signaling pathway of stem-cell-like memory T cells [110], it will be intriguing to see how the Wnt pathway is regulated in T cells expressing CAR that include the CD27 signaling domain. Although much progress has been made in the intracellular signaling domain of CARs, many of these constructs still need to be evaluated in clinical trials.
3.2.2.2 Improving the Human Application of T Cells Expressing CAR
Targeting one TAA that is exclusively expressed on tumor cells is difficult, especially given the heterogeneity of antigen expression on solid tumors. An approach to increase the specificity of genetically modified T cells is to synchronously target two cell surface TAAs expressed on tumors, thereby enabling CAR+ T cells to distinguish tumor cells from normal cells. This can apparently be achieved using two CAR designs which activate T cells via CD3ζ on one CAR and a co-stimulatory molecule (CD28 or CD137) on the other CAR. Kloss et al. developed CARs targeting PSMA and prostate stem cell antigen (PSCA) as model antigens and showed that T cells expressing both anti-PSMA CAR (CD28 + CD137) and low-affinity anti-PSCA CAR (CD3ζ) kill only those tumor cells that express both TAAs [111]. A probable limitation is that the affinity of scFv linked with CD3ζ should be low as described by the authors. The antigen density and affinity of scFv vary with each CAR to tumor combination, and therefore, this needs to be taken into consideration when applying this strategy to the clinical setting.
Autologous T cells are infused in many current clinical trials, but T cells from heavily pre-treated patients may be damaged in quality and compromised in quantity, and (due to the time required for production and manufacture) they may not be available where and when the patient needs them. Any time delay before infusion allows disease progression to take place, and the possibility that treatment is therefore not possible for some patients. “Off-the-shelf” (OTS) therapy administering T cells expressing a CAR would resolve these issues. Toward this end, CAR+ T cells have been pre-prepared from healthy donors, using either peripheral blood mononuclear cells (PBMC) or umbilical cord blood (UCB) derived mononuclear cells, for infusion when the patient needs them rather than when the T cells are available. A potential problem using OTS T cells is the development of an allogeneic immune reaction (either from donor to patient or from patient to donor). Zakezewski et al. generated precursor CAR+ T cells by in vitro differentiation of CAR-transduced HSCs activated and propagated on Notch-1 ligand expressing OP9 cell lines [112]. When precursor CAR+ T cells were infused into irradiated mice they became host MHC restricted and host tolerant. This technology may be used to generate “OTS” CAR+ T cell precursors.
We have shown that the endogenous TCRαβ can be eliminated from CAR+ T cells by ZFNs targeting either TCRα constant region or TCRβ constant region [113]. These TCRαβneg CAR+ T cells can then be enriched by clinically compatible paramagnetic bead-based sorting. TCRαβneg CAR+ T cells cannot proliferate by TCRαβ stimulation while maintaining activity through CAR. We have also shown that HLA can be eliminated from CAR+ T cells, thus enabling these cells to evade attack from patient T cells recognizing allogeneic antigens. Although further evaluation is needed, CAR+ T cells may be used as an OTS “drug” in the growing field of immunotherapy.
3.2.2.3 Clinical Trials Infusing CARs
Multiple phase I clinical trials administering T cells expressing first-generation CARs have been reported [84–87, 114, 115]. Overall clinical responses in these studies are limited presumably due to incomplete T cell activation. To provide additional stimulation to these T cells, Pule et al. reported that half the patients treated with anti-GD2 CAR expressed in EBV-specific T cells showed clinical responses. These EBV-specific T cells presumably retained sufficient stimulation through the EBV-specific TCRαβ to enable targeting of tumor via first-generation CAR. This observation reinforces that co-stimulation is important to improve anti-tumor function.
The results of early-phase clinical studies using second-generation CAR (including CD28 or CD137 intracellular domain) are promising [94, 116–121]. The improved anti-tumor responses appear to correlate with sustained in vivo persistence of CAR+ T cells. For example, Kalos et al. reported that all three patients with advanced chronic lymphocytic leukemia who received CD19-specific T cells expressing a CAR that signaled through chimeric CD137 showed marked clinical responses [117]. It was calculated that on average one CAR+ T cell can act as a serial killer and eradicate 1,000 tumor cells. Most adverse events related to CAR+ T cell infusion were tolerable with intensive clinical support. Expected on-target toxicity occurred as these CAR+ T cells do not distinguish between CD19 on malignant versus normal B cells. Thus, eradication of normal B lineage has served as a biomarker and has been observed. The loss of humoral immunity can lead to complications as one patient has died from viral infection after receiving CD19-specific T cells [119]. However, unexpected on-target related severe adverse events have also reported. In a trial targeting carbonic anhydrase IX (CAIX) CAR T cells were attributed to causing hepatotoxicity due to the low level of CAIX expression on the liver [115]. One patient with metastatic colon cancer treated with ERBB2 target CAR therapy suffered from fatal respiratory distress due to the low level of expression of ERBB2 on lung epithelial cells [122]. Another unforeseen, an adverse event was also reported in a patient with CLL treated with CD19 target CAR. Although the precise cause of death was not reported, this patient showed elevated serum cytokine levels after infusion of CAR+ T cells [123].
3.3 Improving T Cell Function
One of the major challenges in adoptive transfer of genetically engineered T cells is improving T cell function and survival in vivo. One approach to achieving this is to include co-stimulatory signals with the CAR construct. In addition, cytokines known to enhance T cell survival and chemokines that guide T cells to the tumor site(s) have also been tested.
3.3.1 Engineering T Cells to Improve Function and Survival
In vivo survival of infused T cells is important to eradicate residual tumor cells and maintain immune surveillance. BCL2 and BCL-XL are anti-apoptotic molecules that contribute to T cell differentiation and survival [124]. Pro-survival function of cytokines, especially cytokines that signal through the common γ chain, relies on the induction of BCL2 or BCL-XL expression [125]. BCL-XL is also an anti-apoptotic molecule reported to be associated with T cell survival mediated through CD28 co-stimulation [88]. Enforced expression of BCL2 in TAA-specific T cells has been tested in vitro and in vivo [126, 127]. These T cells have augmented resistant to apoptosis, survive in the absence of cytokine support, and exhibit enhanced anti-tumor activity in a mouse model. Genetic modification with BCL-XL protects T cells from Fas-mediated death and enhances survival in vitro and in vivo [127, 128]. Fas-mediated T cell death can be also reduced by introduction of siRNA targeting Fas [129]. hTERT is the catalytic subunit of telomerase and overexpression of hTERT prevents the shortening of a chromosomal ends after multiple cell divisions. Because of this, transduction of hTERT is used for immortalization of cells. hTERT-transduced T cells have been evaluated and found to have enhanced survival [130]. The clinical translation of all these approaches to improve T cell survival needs to be balanced against the risk of infusing a product capable of autonomous proliferation. Thus, at a minimum, these T cells will need to be engineered for conditional ablation through the expression of a suicide gene.
3.3.2 Cytokines to Support T Cell Function and Survival
T cell survival is dependent on factors intrinsic to the T cells and extrinsic as exerted by microenvironment. Cytokines that signal through the common γ chain are indispensable for T cell expansion and survival [131]. These cytokines have been shown to enhance in vivo T cell survival animals and human trials [132–134]. However, the exogenous administration of supraphysiologic concentrations of recombinant human cytokines is expensive, requires repeated injection, and can cause toxicity. Genetic engineering of T cells with enforced expression of cytokines may be an alternative approach to enhancing T cell persistence. The enforced expression of IL-15 has been tested in CD19-specific CAR+ T cells. These T cells secreted IL-15 and proliferated upon encounter with TAA resulting in an improved anti-tumor effect in vivo [135]. However, prolonged expression of IL-15 may lead to genetic alteration and lead to malignant transformation [136]; thus, the co-expression of suicide genes is recommended. IL-7 is an important cytokine for homeostatic expansion of naïve and memory T cells, and IL-7Rα is the important cellular marker that identifies long-lived T cells. However, many T cells lose IL-7Rα after ex vivo culture. The transduction of IL-7Rα into EBV-specific CTL restores responsiveness to IL-7 [137]. Although enforced expression of IL-21 in effector T cells has not been published, IL-21 transduction into T cells capable of antigen presentation (T-APC) has been evaluated. IL-21-secreting T-APCs enhance the generation and proliferation of MART-1-specific CTLs [138]. IL-12 is a heterodimer consisting of a light chain (p35) and a heavy chain (p40) [139]. IL-12 has an important role in T cell priming, effector function, and survival [140]. Several groups have transduced TAA-specific T cells to express IL-12. This resulted in the enhanced cytotoxic function of T cells, but IL-12 alone did not sustain the long-term survival of T cells [141]. Of note, IL-12 expressing CAR T cells acquired resistance to suppression mediated regulatory T cells [142], attracted activated macrophages in tumor site [143], and also reduced tumor infiltration by myeloid suppressor cells [144]. Thus, it appears that IL-12 from T cells has the potential to disrupt a suppressive tumor microenvironment as well as to enhance effector functioning.
3.3.3 Disruption of Inhibitory Molecules
The long-term efficacy of adoptive T cell therapy is subject to immune checkpoint regulation. Interaction of inhibitory molecules on activated T cells and their ligand on tumor cells compromises T cell function. These checkpoints include CTLA-4, PD-1, TIM-3, and LAG3. Such molecules can be targeted by therapeutic mAbs. Promising clinical results infusing mAbs that block the interaction of CTLA-4 with B7.1 (or B7.2) [145, 146] or the interaction between PD-1 and PD-L1 (or PD-L2) [147, 148] in patients with solid tumors have been reported. Combining these mAbs with genetically engineered T cells may enhance their therapeutic efficacy. Alternatively, inhibitory molecules expressed on T cells may also be eliminated by post-transcriptional suppression with shRNA. This has been tested by suppressing PD-1 expression on T cells. Increased production of IFNγ and T cell degranulation was noted although PD-1 suppression was incomplete [149]. With the emergence of gene targeting technologies such as ZFN [150], TALEN [151], and CRISPR/Cas9 [152], these molecules may be targeted to completely disrupt their expression.
3.4 Expression of Chemokine/Chemokine Receptor to Guide T Cells to Desired Sites
In order to completely eradicate tumor cells, intravenously administered T cells need to home to tumor sites. In many types of cancer, the degree of infiltration of T cells at the tumor site, especially memory CD8 T cells, correlates positively with a favorable outcome [2]. Chemokines and their receptors affect the migration of immune cells. Chemokine expression in tumor cells likely guides immune cell infiltration and therefore may alter clinical outcomes [153]. CCL2 chemokine is known to be expressed on many types of cancer cells. Its receptor CCR2 is found on myeloid cells in greater amounts than on T cells. Tumors may defend themselves from T cell attack by producing nitrotyrosine which converts CCL2 on tumor cells to a nitrotyrosinalated CCL2 [154]. Cells expressing high levels of CCR2 can migrate to tumor via the CCR2–CCL2 axis which is co-opted by tumor cells to load their tumor microenvironment with suppressor myeloid cells. The enforced expression of CCR2 on TAA-specific T cells has been tested to improve the trafficking [155, 156]. Such T cells infiltrate into the tumor site at increased efficiency resulting in augmented antitumor activity. Also, the enforced expression of CCR4 in CD30-specific CAR+ T cells enhances migration to sites of Hodgkin lymphoma [157]. The strategy to express chemokine or its receptor on T cells can be generalized by profiling chemokine expression on target tumor cells and enforced expression of corresponding ligand on TAA-specific T cells. Transient expression of chemokine may be sufficient to guide T cells to a tumor site, and therefore, the introduction of in vitro-transcribed mRNA coding for chemokine receptor may be desirable.
3.5 Control of Aberrant T Cell Activity
Suicide gene therapy was first adapted for clinical use in the treatment for cancer. Targeted transduction of a suicide gene into tumor cells was used to destroy tumor by subsequent administration of substrate. This technology was then adapted to enhance the safety of donor leukocyte infusion (DLI) after allogeneic HSCT. Although DLI may induce remission in certain types of hematological malignancies, it can result in life-threatening GVHD. These transgenes capable of conditional ablation were introduced to eliminate genetically modified T cells severe GVHD occurred after DLI. This technology can also be applied to eliminate any type of cells or transformed cells which are potentially harmful to the patient.
3.5.1 Conditional T Cell Ablation Mediated by Kinases
Thymidine kinase is involved in the pyrimidine salvage pathway. Herpes simplex virus type 1 thymidine kinase (HSV-TK) is desirable for gene therapy because of its broad substrate specificity (e.g., pyrimidines, pyrimidine analogs, and purine analogs). Of these potential substrates, ganciclovir (GCV) is favored for TK-mediated suicide gene therapy because of the need for a low drug concentration and that it is clinically available. The administration of GCV leads to DNA chain termination and cell death in cells expressing HSV-TK. The first use for the HSV-TK gene in T cells was to control GVHD after DLI in allogeneic HSCT [158]. Since this first study, hundreds of patients have been treated using this technology in phases I–II studies. Control of GVHD was observed in all patients who developed GVHD [159]. The ability to control GVHD with HSV-TK has also been proved in the more challenging setting of HLA-haploidentical transplantation [160]. In addition to use as a suicide gene, HSV-TK can be used for noninvasive imaging. T cells expressing variants of HSV-TK can be tracked by injection of radiolabeled 2′-fluoro-2′-deoxy-1-beta-D-arabinofuranosyl-5-iodouracil (FIAU) as imaged by positron emission tomography [161]. This is achieved by the trapping of FIAU in the T cell cytoplasm after phosphorylation by HSV-TK [162]. A drawback to HSV-TK is its immunogenicity and the myelosuppression due to GCV infusion. Mutants of HSV-TK have been developed to overcome GCV mediated myelotoxicity. These need lower concentrations of GCV than wild-type HSV-TK gene, thus reducing the potential for myelosuppression [163, 164]. Immunogenicity of HSV-TK leads to the induction of HSV-TK-specific T cells in the host which compromises persistence of the infused genetically modified T cells [165]. A variant of the less immunogenic human-derived thymidine monophosphate kinase (TMPK) can also be used to conditionally ablate T cells upon addition of 3′-azido-3′-deoxythymidine (AZT) [166]. One concern is the amount of AZT that is needed to achieve ablation of T cells if used in humans.
3.5.2 Conditional T Cell Ablation Mediated by Dimerization
Another class of suicide genes is based on the use of a small molecule to dimerize recombinant Fas. This uses a variant of FK506-binding protein (FKBP) fused to Fas and AP1903 that is a chemical inducer of dimerization (CID). T cells expressing this fusion can be conditionally eliminated by administration of CID as a result of cross-linked Fas initiating caspase-dependent apoptosis. This construct has been tested in human [136] and macaque T cells [137]. The extent of drug-mediated T cell death was comparable to that of HSV-TK-transduced T cells (up to 90 %), but the pace of inducing apoptosis in this system was improved. Moreover, because this system is derived from human proteins, this fusion transgene may avoid the immunogenicity of HSV-TK. Straathof et al. generated an alternative using caspase 9 instead of Fas intracellular domain [135]. Several constructs were evaluated, and the construct of one copy of FKBP fused to large and small subunits of caspase 9 (referred to as iCasp9) was preferred. A clinically appealing approach has been developed to generate T cells that all express the iCasp9 and thus are all susceptible to conditional ablation. This was based on co-expression of iCasp9 with recombinant CD19 and using paramagnetic beads to positively select successfully genetically modified T cells with mAb against CD19. The initial clinical reports infused haploidentical CD19+ (iCasp9+) T cells after HSCT [136]. Four out of 5 patients infused with transduced T cells developed GVHD. Their GVHD resolved after one infusion of CID due to the rapid elimination of the genetically modified transduced T cells. Although further clinical experience with this suicide gene therapy is necessary, iCasp9 may be the best alternative to HSV-TK.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


