Species
Peak wavelength (nm)
Size (kDa)
Native substrate
Co-factors
Native subcellular localization
Reference
Photinus pyralis
560
62
D-luciferin
ATP, O2
Intracellular (peroxisomes)
[91]
Phrixothrix hirtus
622
61
D-luciferin
ATP, O2
Intracellular (peroxisomes)
[92]
Pyrophorus plagiophthalamus
593
60
D-luciferin
ATP, O2
Intracellular (peroxisomes)
[93]
Renilla reniformis
480
36
Coelenterazine
O2
Intracellular (lumisomes)
[94]
Gaussia princeps
470
20
Coelenterazine
O2
Secreted
[95]
Metridia longa
480
24
Coelenterazine
O2
Secreted
[96]
Oplophorus gracilirostris
455
106 (heterotetramer)
Coelenterazine
O2
Secreted
[97]
Vargula hilgendorfii
460
68
Cypridina luciferin
O2
Secreted
[98]
Cypridina noctiluca
465
61
Cypridina luciferin
O2
Secreted
[18]
Pyrocystis lunula
474
140
Dinoflagellate luciferin
ATP, O2
Intracellular (microsources)
[19]
Vibrio harveyi
490
77 (heterodimer)
RCHO
FMNH2, O2
Intracellular
[20]
8.2.1 Beetle Luciferases
Terrestrial organisms have included luciferases in their evolutionary trajectory for a wide variety of applications, including defense, courtship, and the attraction of prey. Within the terrestrial luciferases, the best studied and by far the most commonly used in biomedical applications are the “beetle luciferases” found in organisms within the order Elateroidea. Although many beetle luciferases have been identified, including those from the click beetle (Pyrophorus plagiophthalamus) and the railroad worm (Phrixothrix hirtus), the most well-known and widely studied is that from the North American firefly, Photinus pyralis. The P. pyralis luciferase (generally referred to as firefly luciferase = FLuc) is a 62-kDa globular protein and can be thought of as the prototype for the beetle luciferases. The beetle luciferases are homologous, consistent with them evolving only once during the history of this planet. They all catalyze the identical enzymatic reaction, producing light by oxidizing their substrate D-luciferin in the presence of cofactors Mg2+, O2, and ATP, but they can demonstrate slight differences in their emission wavelength. The beetle luciferases have been the focus of many engineering pursuits, mainly due to their earlier discovery, relatively redshifted emission compared to other well-studied luciferases, and their necessity for ATP, a central element in cellular metabolism, as a cofactor.
The oxidation of D-luciferin by beetle luciferases is a two-step enzymatic process (Fig. 8.1) [5]. The first step involves the reaction of luciferin and ATP to form luciferyl-adenylate. This step is similar to the one performed by many fatty acyl-CoA synthetases, and some evidence suggests that beetle luciferases are evolutionarily connected to these enzymes [6]. The second step of the reaction involves oxidation of luciferyl-adenylate by molecular oxygen to form an intermediate structure, which spontaneously breaks down into oxyluciferin and carbon dioxide. The energy released by the destruction of the chemical bond takes the form of a photon, and thus bioluminescence is produced.
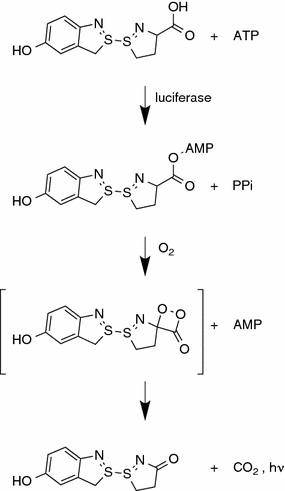
Fig. 8.1
The oxidation of luciferin by beetle luciferases is a two-step process that is initiated by the formation of a luciferyl-adenylate intermediate. This chemical is then oxidized in the presence of molecular oxygen and light is generated as a by-product
8.2.2 Coelenterazine Luciferases
A variety of luciferin/luciferase systems also exist in marine environments. These marine luciferases most commonly emit light with a blue wavelength peak (~480 nm), likely reflecting the end result of selective pressure originating from the preferential transmittance of blue light by ocean water [7]. Within the marine luciferases, the best studied and the only ones in widespread biomedical utilization are those that utilize the substrate coelenterazine (or analogs of coelenterazine).
Coelenterazine is an imidazolopyrazine common in the marine food chain. In contrast to the beetle luciferases, coelenterazine-utilizing luciferases have evolved multiple times from a variety of different precursor enzymes to utilize this same substrate. This likely reflects the relatively simple chemistry and low kinetic barrier of the coelenterazine degradation scheme in comparison to D-luciferin. Although some specific coelenterazine luciferases may require cofactors, in general, these luciferases require only coelenterazine and oxygen, producing coelenteramide, carbon dioxide, and a blue wavelength photon of light. The ease with which a new enzyme can arise that catalyzes this reaction is exemplified by the observation that even albumin catalyzes the degradation of coelenterazine at a low level [8]. This multiplicity is advantageous for protein engineering as there exists a number of very different starting points for developing novel luciferases.
The well-known coelenterazine luciferases can be categorized into three classes, luciferases from the genus Renilla (e.g., Renilla reniformis luciferase), copepod luciferases such as those from the family Metridinidae (e.g., Gaussia princeps, Metridia longa), and more recently the luciferase from the decapod Oplophorus gracilirostris. These different luciferases also demonstrate variable levels of specificity to coelenterazine versus analogs of coelenterazine, with G. princeps luciferase being the most specific, and O. gracilirostris luciferase being the least [9].
The coelenterazine luciferase most widely studied is that from the soft coral R. reniformis. This luciferase is often simply called “Renilla luciferase” (RLuc), although there are other species within the Renilla genus that have had their luciferases isolated (e.g., Renilla mülleri luciferase). Renilla luciferase is a 36-kDa intracellular monomeric protein that is efficiently expressed in a variety of bacterial and mammalian expression systems [10].
The second most studied coelenterazine luciferase is the luciferase from the copepod G. princeps, generally referred to simply as Gaussia luciferase (GLuc). GLuc is a 20-kDa protein, and similarly to the other copepod luciferases, is a secreted protein that harbors multiple disulfide bonds. These disulfide bonds have made copepod luciferases challenging to express in functional form in bacterial expression systems, although success has been obtained with insect [11] and cell-free systems [12]. The primary sequence of copepod luciferases contains two similar functional domains [13]. In the case of GLuc, these domains have demonstrated an ability to catalyze coelenterazine degradation independently, albeit with greatly decreased activity [14]. A potential limitation of the copepod luciferases to keep in mind is that their enzymatic action appears to be cooperative [15] and therefore light output is nonlinearly related to substrate concentration. However, in most assays, substrate concentration is relatively constant and luciferase concentration is the variable being measured, so this positive cooperative effect is rarely noticeable.
The coelenterazine luciferase from the decapod O. gracilirostris (OLuc) is a secreted enzyme complex, consisting of a heteromeric structure containing two 35-kDa and two 19-kDa catalytic subunits [16]. This complexity has previously limited its application to biomedical research. Recently, a monomeric luciferase (termed NanoLuc) has been derived by protein engineering from the 19-kDa OLuc catalytic subunit [17]. This OLuc variant should be more applicable to biomedical applications and is described in more detail later in this chapter.
8.2.3 Other Marine Luciferases
Additional marine luciferases have been studied, but have found much more limited use in biological assays and have not been extensively engineered. Two luciferases from Cypridina ostracods have been cloned, one from Vargula hilgendorfii, and one from Cypridina noctiluca. These Cypridina luciferases emit blue light when degrading their substrate Vargulin. Given their unique substrate, they can be multiplexed in experimental use with D-luciferin and/or coelenterazine-utilizing luciferases. Although highly stable, these proteins are relatively large (61 kDa), are secreted enzymes, contain a total of 17 disulfide bonds [18], and their substrate vargulin is relatively unstable, all factors that would make the Cypridina luciferases a more challenging starting point for protein engineering than the coelenterazine or Beetle luciferases.
Several marine dinoflagellates express a luciferase, most likely used for quorum sensing. This luciferase is relatively large at ~140 kDa, and emits blue wavelengths. These properties have prevented a wide adoption of this luciferase, but its unique substrate provides the possibility for multiplexing with other luciferases. A 46-kDa active fragment of dinoflagellate luciferase has been developed and utilized as a reporter in mammalian cells [19]. This truncated dinoflagellate luciferase could be combined with firefly and Renilla luciferases, for example, to monitor the expression of three different genes.
8.2.4 Bacterial Luciferase
Luciferase-expressing bacteria can be found in a variety of ecosystems. Most possess symbiotic relationships with other organisms, providing a means for communication, attraction of prey, or defense to the host in exchange for nutrients. The bacterial luciferase is actually a cassette of five genes, two of which are responsible for substrate oxidation leading to bioluminescence, and three that synthesize the substrate from common biomolecules. Due to this level of complexity, the bacterial luciferase is seldom used in biomedical research, although efforts have been made to adapt it for the creation of autonomously luminescent cells [20]. This achievement is attractive for certain longitudinal studies of specific cellular populations because it obviates the necessity for repeated administration of exogenous luciferase substrate.
8.3 Current Biomedical Uses
Due to the intrinsic difficulties of translating optical technologies into a clinical setting (i.e., suboptimal wavelengths, necessity for exogenous genetic/proteinaceous material), luciferases have a limited role in the medical setting. They are widely used, however, in biomedical research, and are gaining ground in the area of diagnostic testing. One technique in particular named luciferase immunoprecipitation system (LIPS) has demonstrated considerable advantages over similar systems that use fluorescent or colorimetric indicators. The LIPS procedure is very similar to enzyme-linked immunosorbent assay (ELISA), with the exception that an antibody-luciferase fusion protein is used for the detection of antigens in assays. Compared to similar immunodetection procedures, LIPS has proven to be more sensitive and specific [21]. As with other applications that will be discussed below, the use of luciferases for immunodiagnostics requires that the enzymes maintain stability over time and in some cases at elevated temperatures. Thus, engineering thermostable luciferases is crucial for the success of these techniques.
One of the biggest commercial successes of the beetle luciferases is pyrosequencing. This is a DNA sequencing technique that is predicated on three enzymatic reactions. The first reaction occurs during de novo DNA synthesis by DNA polymerase. This reaction produces an inorganic pyrophosphate molecule, which can then be converted into ATP by ATP sulfurylase (the second reaction). The final reaction is described in more detail in the previous section and involves the use of this ATP molecule by a beetle luciferase to produce a bioluminescent signal. Therefore, each nucleotide successfully integrated into the growing complementary chain of the template DNA can be quantitatively detected by light emission. Pyrosequencing has been shown to be quicker and simpler than other sequencing techniques and has become the method of choice for many sequencing needs [22]. As DNA polymerization is generally optimal at elevated temperatures, thermostable luciferases are absolutely necessary for this application, and many luciferase engineering studies have been performed to produce more pyrosequencing-friendly beetle luciferase variants.
Additionally, luciferases are used extensively in basic laboratory biomedical research as reporter genes. Traditionally, certain genes whose protein products are detectable by an optical or radiological device (e.g., green fluorescent protein, beta-galactosidase, thymidine kinase) have been used as surrogates to report on a genetic activity of interest. Often this takes the form of driving the expression of a reporter gene with the promoter of the gene of interest. In this manner, the transcriptional activation of a gene can be interrogated using convenient and fast readouts. Luciferases have proven to hold a special niche in this regard as they are often more sensitive than other reporter genes and generally translate better to small-animal in vivo studies. Moreover, due to the technique known as split-protein complementation, discussed in greater detail in a subsequent section, luciferases have been engineered to report on much more than gene expression. Recent studies have seen the design of luciferases that can report on small molecule kinase inhibition, DNA methylation, and caspase activity [23–25]. These studies require luciferases that have been specially engineered to only produce bioluminescence after these molecular events have occurred and demonstrate the degree of creativity and ingenuity currently being applied to luciferase engineering.
Small-animal in vivo imaging represents the final main application of engineered luciferases. This application is in many ways an extension of reporter gene assays, but imposes additional constraints upon the characteristics of the luciferase used. It generally favors the use of luciferases whose wavelengths have been shifted toward the red end of the electromagnetic spectrum due to the optical window of mammalian tissue (discussed in greater detail in a subsequent section). These engineered luciferases have contributed greatly to our ability to translate cellular studies into more physiologically relevant models and have provided a potent tool for interrogating biological processes in a living system.
8.4 Rationale for Protein Engineering of Luciferases
Although there are a number of reasons for performing luciferase protein engineering, a common reason is that the native luciferin/luciferase system is not sufficiently robust for consistent use in a variety of laboratory solutions, cellular compartments, or temperatures. Each luciferin/luciferase system has evolved under the selective pressures of its native environment. For instance, Renilla luciferase evolved to work in a salt water creature, in small membrane-bound particles called lumisomes, associated with a green fluorescent protein, and at ocean temperatures [26]. Thus, it stands to reason that mutations could be readily identified to improve its performance and stability as a monomeric protein in laboratory solutions at room or mammalian body temperature.
Additionally, luciferases are relatively “easy” to engineer compared to other enzymes because their functional properties make mutants readily identifiable. Large-scale assays can be performed utilizing common bioluminescence imaging systems, or even the human eye for sufficiently bright luciferases [27].
8.5 Designing Selection Strategies for Protein Engineering of Luciferases
An important philosophical point in the protein engineering of luciferases is the definition of what constitutes “better.” Much as beauty is in the eye of the beholder, what constitutes a “better” luciferase is entirely dependent on the end application goal. Most comparisons between luciferases in the literature are made with respect to which is “brighter” in a particular assay condition, with little or no consideration given for the robustness of the improvement when the assay is performed in different solutions/cellular compartments/temperatures or the actual enzyme kinetics. For instance, a kinetically slow enzyme could look “better” than a fast enzyme if the fast enzyme degrades the majority of the applied luciferin before brightness is measured. A secreted luciferase may perform “better” than a nonsecreted luciferase in the context of a cell culture when the entire cell culture dish including media is assessed, but the loss of association between the cell of origin and the location of bioluminescence will limit utility of a secreted luciferase in small-animal bioluminescence imaging.
A closely related point is that many comparisons are based on assays (such as cell culture assays) in which the bioluminescence signal is not normalized to the total amount of luciferase. This gives an advantage to luciferases that are more stable, as more of the luciferase will accumulate over the course of the experiment, and the experimental condition utilizing the more stable luciferase will appear to be “brighter” even though both luciferases may have the same light output per enzyme. Given the end goal of the assay, this stability may or may not be desired. Increased luciferase stability impedes the ability of an assay to monitor transient fluctuations in luciferase expression, and it takes longer to reach a steady state of luciferase activity within the cell. As discussed in a following section, some luciferase engineering has been directed into making the luciferase mRNA and/or protein less stable for this very reason.
This all leads to the first law of directed evolution: “you get what you select for.” The improved luciferase/luciferin combinations derived under artificial selective forces in the laboratory are being improved with respect to activity in a specific test. Screens must be designed carefully, or contain controls, to ensure that the mutations picked up are being selected for the desired end application goals. As an example, consider a random mutagenesis screen of a luciferase in E. coli. If the selection method is simply which colonies are brightest the screen will select for a number of different properties: increased thermostability at the incubation temperature, improved codon utilization for bacterial expression, removal of bacterial protease sites, improved folding in the bacterial expression system, better matching of the emission spectrum to the sensitivity profile of the detector, etc. Some of these “improvements” may be detrimental if the luciferase is utilized in a different system such as mammalian cells. If a more targeted goal is desired, such as faster enzyme kinetics, a more sophisticated screening system must be employed.
8.6 Methods for Protein Engineering of Luciferases
The techniques and theories driving luciferase engineering are not substantially different from those used to modify any other protein, but luciferase’s functional capacity has made it a facile model for experimentation, and its prominence in biomedical research has made it an attractive target.
Luciferase engineering can be broadly divided into two categories. The first category is similar to canonical protein engineering, in which changes made to the primary sequence affect the enzyme’s intrinsic properties (e.g., specific activity, thermostability, emission wavelength).
The second category is more prominent, although not exclusive, to reporter proteins such as the luciferases, and involves the dissection or interruption of the protein’s primary sequence into two distinct domains. The purpose of alterations in this second category is to prevent the enzyme from oxidizing its substrate in the absence of some other biomolecule, thus serving as an analyte detector.
Within each category, various strategies have been employed to introduce mutations. Random mutagenesis has been applied to luciferase engineering with substantial success [28–33]. Different techniques have been used to induce random mutations, although the most common is error-prone PCR. In theory, this approach yields an unbiased distribution of mutants across the coding region of the protein, effectively maximizing the variable space. In practice, certain codons are more susceptible to mutagenesis than others, and the altered residues are often less randomly distributed than expected. Nevertheless, this technique yields a prodigious number of mutated enzymes that can be subjected to high throughput screens. In the case of luciferase engineering, the screens generally take the form of bioluminescent output under various constraints. For example, one screen used by several investigators to identify thermostable variants involves the incubation of luciferase-expressing bacterial colonies at an elevated temperature followed by application of the luminescent substrate and identification of the brightest colonies. Constructs are then sequenced to identify the mutations contributing to the new phenotype.
The other main technique for introducing mutations into luciferase is site-directed mutagenesis. Unlike random mutagenesis, this method is usually based on a specific hypothesis regarding how a certain mutation or mutation site will affect the enzyme’s properties [10]. Site-directed mutations are most often initiated by structural or sequence analysis and can be far more efficient than random approaches. For instance, in the generation of the RLuc variant RLuc8 [10], only ~30 single-mutation RLuc variants were screened to generate a variant that was fourfold brighter and two orders of magnitude more stable.
Combinatorial mutagenesis is a variation on both of these methods for introducing mutations. This semi-rational method entails the random incorporation of a subset of site-specific mutations. Although this method has been used sparingly for luciferase engineering [28], its results are encouraging and may see wider use in the future.
8.6.1 Structure-Based Versus Sequence-Based
Sequence homology between the luciferase proteins of various species has driven many of the studies that seek to enhance these enzymes’ properties. This has been a motivating impulse for those seeking to induce bathochromatic shifts in the beetle luciferases, as well as groups interested in improving protein stability in both beetle and coelenterazine luciferases, although the precise rationale in each case is slightly different. To provoke changes in peak wavelength of the beetle luciferases, sequence homology is often assessed in an attempt to graft the attributes of one luciferase onto another.
Sequence homology has also been used with great success for the creation of more stable luciferase enzymes [10, 34, 35], although for this application, homology is assessed between multiple species. One of the guiding principles of protein evolution is that conservation is driven either by stability or function. By selectively mutating residues within a luciferase so that they conform to a consensus sequence, one is choosing candidate mutations that have already been screened by nature to be tolerated or even preferred within the context of the proteins’ fold and are much less likely to be deleterious to the protein than a residue picked at random. This consensus mutagenesis approach requires that a number of homologous proteins already exist in the sequence database that, while being similar enough to allow a valid alignment, are evolutionarily distinct enough that a bias toward stabilizing mutations can be identified.
Structural considerations have also played a role in luciferase engineering. Different tactics have been used in this respect, including mutagenesis of residues involved in the active site, introduction of cysteine residues to form disulfide bridges, and mutagenesis of solvent-exposed residues [28, 36–38]. These studies use publicly available crystal structures of a handful of luciferases to develop biochemical hypotheses regarding the contribution of individual amino acids to stability and wavelength emission.
8.6.2 Codon Optimization
Although not strictly protein engineering as the primary sequence of the protein is conserved, evaluation of codon usage is often a useful initial step in improving expression levels of luciferase proteins. Several studies have generated codon-optimized luciferase genes, primarily for mammalian expression, to help improve transcriptional activity and mRNA stability [39–42]. This procedure is not specific for luciferases and is generally performed when attempting to generate robust expression of a nonmammalian gene in a mammalian system.
8.6.3 Protein Truncation/Extension
Certain studies have manipulated the stability or intracellular compartmentalization of luciferases by truncating or appending additional residues to the primary sequence. Again, this technique is not specific to luciferase engineering and has been applied to many other proteins.
In general, these modifications are used to alter properties of the luciferase that are of importance when used as a reporter gene (discussed in further detail later). Adding the PEST sequence or ubiquitinylation-prone sequences to the luciferase protein has the general effect of reporting on cellular dynamics and intracellular protein transport [43–45]. Removal of N-terminal signal peptide sequences or adding transmembrane domains have been utilized to maintain association of a normally secreted protein to its cell of origin [41, 46].
8.6.4 Substrate Alteration
An in-depth discussion of chemical modifications to luciferase substrates, chiefly D-luciferin and coelenterazine, is beyond the scope of a chapter focused on protein engineering of luciferases. However, it is important to note that an equally large body of work has reported on the successful design of modified luciferase substrates that have the potential to alter emission wavelength when oxidized, or prevent oxidation prior to conversion by a molecule of interest. Thus, equivalent results in terms of red shifts and molecular-sensing have been achieved by engineering of the luciferase substrates rather than the luciferases themselves [28, 47, 48].
It bears mentioning, that a factor to consider when comparing between different substrates is the autoluminescence rate of the luciferin under the assay conditions employed. For instance, the coelenterazine analog coelenterazine-v (Fig. 8.2) generates an order of magnitude higher background autoluminescence than coelenterazine [47]. Even if a luciferase is brighter with coelenterazine-v, the increase in brightness is unlikely to make up for the order of magnitude decrease in sensitivity for conditions in which very small amounts of luciferase are present.
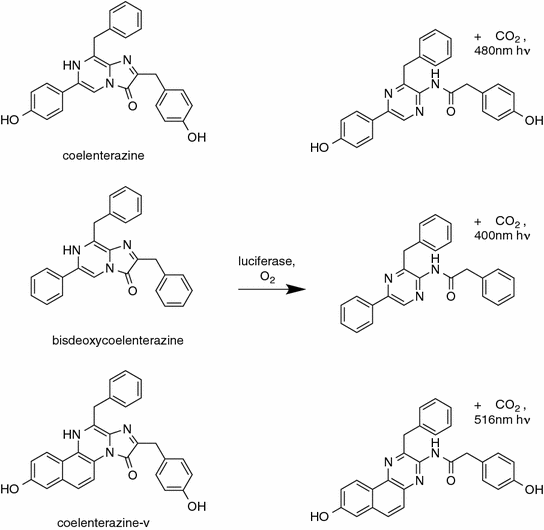
8.7 Examples of Luciferase Protein Engineering
8.7.1 Intensity Improvements
Although only a single study in the literature has specifically sought to enhance the intensity of beetle luciferase, many mutants have demonstrated increased light output as a result of stabilizing or color-shifting mutations. Further complicating the issue of comparing mutants between studies is the number of variables involved in measuring luminescent output (e.g., luciferin concentration, ATP concentration, acquisition time, photomultiplier tube wavelength sensitivity). For the purposes of this chapter, activity values relative to the mutant’s parenteral luciferase are reported when possible, as this accounts for variations in luminescence measurements. Using this metric, the brightest, or most intense, mutant described to date is a triple mutant of the P. pyralis luciferase (I423L, D436G, L530R), which demonstrated a 12-fold increase in intensity over wild type [32]. Each mutation was discovered independently by random mutagenesis coupled with screening of bacterial cell lysates, and then all three mutations were combined to produce the final version. Also of note in this category of engineered luciferases is a report of a double mutant of the P. hirtus luciferase (I212L, N351K), yielding a 9.8-fold improvement in intensity over wild type [34]. These amino acid alterations were introduced by site-specific mutagenesis based on sequence homology to stabilizing mutations previously characterized in the luciferases of Luciola cruciata and P. pyralis. While even this mutant is much less bright compared to its compatriots in the luciferase gallery due to the low intrinsic activity of wild-type P. hirtus, this study is of particular interest because of the long peak emission wavelength of P. hirtus, an attribute which is examined in the following section.
Similar to the case for beetle luciferases, while a number of studies have generated “improved” coelenterazine luciferases, improvements in light output have generally been due to improvements in stability and therefore the amount of functional protein, rather than mutations that improve the quantum yield or kinetics of the enzyme. Utilizing a consensus sequence approach, an M185V mutation was identified for Renilla luciferase that led to a threefold increase in light output arising from improvements in both quantum yield and kinetics [10]. A random mutagenesis screen identified the mutations K189V and V267I that led to a threefold to fourfold improvement in light output compared to native Renilla luciferase [49]. These improvements were attributed to a combination of improved stability, improved kinetics, and decreased levels of substrate inhibition. These mutation sites had previously been studied with other amino acid substitutions based either on a consensus mutation approach (V267) or due to their proximity to the active pocket (K189), but these other substitutions at the same sites did not yield improvements [27].
For the copepod luciferases, recognition that the N-terminal domain of M. longa was not homologous to Gaussia luciferase led to an N-terminal-truncated M. longa variant that demonstrated sixfold to tenfold improvements in light output compared to the native luciferase, albeit with decreased thermal stability [50]. A semi-rational mutagenesis strategy was performed on Gaussia luciferase by targeting hydrophobic regions presumed to constitute the enzymatic pocket, and substituting in other hydrophobic amino acids [51]. Screening of the resultant single-mutation variants and combining the beneficial mutations, led to a 4-mutation variant termed “Monsta” that exhibited ~six-fold greater light output than GLuc, with this improvement due to improved folding, turnover rate and quantum yield.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


