Years
Authors
Area of PPI study
Imaging assay
Reporter used
References
2002
Ray et al.
Development of new assay
Two-hybrid system
Fluc
[68]
2002
Luker et al.
Development of new assay
Two-hybrid system
TK and Fluc
[69]
2002
Paulmurugan et al.
Development of new assay
Split reporter complementation and reconstitution
Fluc
[31]
2003
Luker et al.
PPI between p53 and large T antigen of SV40 virus
Two-hybrid system
TK and Fluc
[70]
2004
Paulmurugan et al.
Rapamycin modulation of FRB and FKBP12 PPI
Split reporter complementation
Rluc
[55]
2004
Massoud et al.
Homodimeric PPIs
Split reporter complementation
Rluc
[28]
2004
Kim et al.
Protein nuclear transport
Split reporter reconstitution
Rluc
[45]
2004
Luker et al.
Development and applications of new split reporter fragments
Split reporter complementation
Fluc
[22]
2005
Paulmurugan et al.
Development of self-complementing split reporter fragments
Split reporter complementation
Fluc
[54]
2005
Paulmurugan et al.
Development of a fusion protein approach to image drug modulation of PPIs
Split reporter complementation
Rluc
[58]
2005
De et al.
Development of a new assay
BRET
Rluc (and GFP variant)
[71]
2005
Kim et al.
Detection of stress-related corticosterone level increases
Split reporter reconstitution
Rluc
[46]
2006
Kanno et al.
Detection of protein release from mitochondria to cytosol
Split reporter reconstitution
Rluc
[47]
2007
De et al.
Further developments of an assay
BRET
Rluc (and GFP variant)
[72]
2007
Zhang et al.
Detection of Akt kinase activity
Split reporter complementation
Fluc
[52]
2007
Paulmurugan et al.
Detection of mutiprotein PPIs
Split reporter complementation
Fluc and Rluc
in text
2008
Choi et al.
PPI between HIF-1 alpha and VHL
Split reporter complementation
Fluc
[50]
2008
Chan et al.
Detection of HSP90 inhibitors
Split reporter complementation
Rluc
[59]
2008
Zhang et al.
Enhanced detection of Akt kinase activity
Split reporter complementation
Fluc
[53]
2008
Luker et al.
Activation of the chemokine receptor CXCR4
Split reporter complementation
Fluc
[73]
2008
Pichler et al.
Development of a universal transgenic reporter mouse for PPI detection
Two-hybrid system
Fluc
[74]
2009
De et al.
Further developments of an assay
BRET
Rluc (and GFP variant)
[75]
2009
Chan et al.
Detection of protein phosphorylation mediated by protein kinases
Split reporter complementation
Fluc
[51]
2010
Pan et al.
Monitoring of rapamycin pharmacodynamics
Two-hybrid system
Fluc
[76]
2010
Massoud et al.
Development of a PET-based split reporter
Split reporter complementation
TK
[65]
2011
Dragulescu-Andrasi et al.
Detection of deep seated PPIs
BRET
Rluc
[77]
Several areas of investigation are required to further refine the use of the modified mammalian two-hybrid system for noninvasive imaging of PPIs confined to the nucleus. As well as further quantitative and kinetic evaluations (e.g., characterizing the ability to follow interactions over time based on the half-life of the reporter protein(s) that are transactivated), studies are needed to optimize the choice of transactivator as well as the choice of promoters and levels of fusion proteins. This strategy will not be discussed further in this chapter; and a separate chapter dedicated to BRET imaging can be found elsewhere in this book.
In certain circumstances, functional proteins can assemble from one or more polypeptide fragments, with the occurrence and efficiency of assembly commandeered into a strategy to measure real-time PPIs. Indeed, synthetically separated fragments of some enzymes can reconstitute functionally active protein particularly if the interaction is helped along by fusion of the enzyme fragments to strongly interacting moieties. Thus, in the “split protein” strategy, a single reporter protein/enzyme is cleaved into N-terminal and C-terminal segments; each segment is fused to one of two interacting proteins (X and Y). Physical interactions between the two proteins X and Y reconstitutes the functional reporter protein leading to signal generation that can be measured. This split protein strategy can work either through protein-fragment complementation assays (PCA), or intein-mediated reconstitution assays. In the former, noncovalent assembly of the reporter protein occurs, and in the latter case, reconstitution of the full reporter protein occurs through covalent bonding. To date, several reporter proteins (e.g., β-lactamase, β-galactosidase, ubiquitin, dihydrofolate reductase, firefly luciferase [Fluc], Renilla luciferase [Rluc], Gaussia luciferase [GLuc], Click beetle luciferase [CBLuc], green fluorescent protein) have been adapted for split protein strategies by finding various split sites for each reporter protein [29–32]. If a full-length reporter can be imaged in living subjects, and this reporter can be appropriately split, then the split reporter assay could possibly be used to noninvasively image PPIs. The appropriate split point should lead to two fragments that do not have significant affinity for each other and yet when brought together (through interaction of the two proteins being studied for their mutual affinity) lead to detectable signal.
The principle of the PCA strategy for detecting PPIs was first demonstrated by Pelletier et al. using the enzyme dihydrofolate reductase (DHFR) [33], following inspiration from a 1994 paper by Johnsson and Varshavsky [34] describing what they called the “ubiquitin split protein sensor.” In all PCAs, splitting a specific reporter protein into two distinct fragments abolishes its function. Bringing the two fragments back together in a controlled manner then restores functional activity [35]. Selected fragments of many proteins can associate to produce functional bimolecular complexes [36]; the PCA system can therefore be generalized for a number of enzymes for the detection of PPIs, examples including DHFR, glycinamide ribonucleotide (GAR) transformylase, aminoglycoside, and hygromycin B phosphotransferases, all reviewed by Michnick et al. [35], Escherichia coli TEM-1 β-lactamase [29, 37], green fluorescent protein and its variants [36], and the molecular imaging reporters Fluc [31] and Rluc [30, 38]. A review of this subject was also published recently by Shekhawat and Gosh [39].
9.4.1 Intein-mediated Reconstitution Assays
These assays are based on the restoration of the full reporter protein through covalent bonding. Inteins have been defined as protein sequences embedded in-frame within a precursor protein sequence and excised during a maturation process termed protein splicing [40]. Protein splicing is a post-translational event involving precise excision of the intein sequence and concomitant ligation of the flanking sequences (N- and C-exteins) by a normal peptide bond [41]. Inteins are intervening DNA sequences that are not present in the mature gene product as a result of splicing at the protein level instead of at the RNA level. In 1998, it was discovered that the gene for the catalytic α subunit of the replicative DNA polymerase III from Synechocystis sp. PCC6803 (Ssp) is encoded in two segments dnaE–n and dnaE–c [42]. Inteins represent a potentially powerful means of protein manipulation, because two peptide bonds are broken, and a new peptide bond is formed during the protein splicing process. Protein splicing is an exceedingly complex self-catalyzed process that requires neither cofactors nor auxiliary enzymes. It requires no source of metabolic energy and therefore involves bond rearrangements rather than bond cleavage followed by re-synthesis. The elucidation of the reaction steps involved in protein splicing has made it possible to modulate the reactions by mutations and to design proteins that can undergo highly specific self-cleavage and protein ligation reactions. An intein can be viewed as an enzyme whose substrate is the adjacent amino acid residues in the two exteins to which it is linked.
Ozawa et al. [43] initially demonstrated that Fluc can be split between amino acid positions 437 and 438 and used with inteins (DnaE) in a reconstitution strategy to detect insulin-induced interaction of phosphorylated insulin receptor substrate 1 (IRS-1) and its target N-terminal SH2 domain of phosphoinositide-3 kinase (PI-3 K) in a cell culture assay. Upon interaction of the two proteins, the two DnaE fragments are brought close enough to fold together and initiate splicing and linking of the two Fluc halves with a peptide bond. The Fluc gene has to be rationally dissected so that each half of Fluc is inactive. After ligating the Fluc fragments together, the resultant mature Fluc recovers its bioluminescence activity [44].
We subsequently reasoned that it may be possible to split Fluc and use split reporter complementation without inteins. We therefore studied PCA and intein-mediated reconstitution of Fluc fragments and found that a complementation strategy was as sensitive as the intein-mediated reconstitution strategy under the conditions tested [31]. Thus, we demonstrated for the first time the feasibility of imaging PPIs using split reporters in small living animals. We studied a PCA based on split Fluc (cleaved into two fragments nFluc: residues 1–437; and cFluc: residues 438–550), using the interaction of Id and MyoD as test proteins [31].
Subsequently, Kim et al. [45] developed a genetically encoded bioluminescence indicator for monitoring and imaging the nuclear trafficking of target proteins in vitro and in vivo. The principle is based on reconstitution of split fragments of Rluc by protein splicing with a DnaE intein. A target cytosolic protein fused to the N-terminal half of Rluc is expressed in mammalian cells. If the protein translocates into the nucleus, the Rluc moiety meets the C-terminal half of Rluc, and full-length Rluc is reconstituted by protein splicing. They demonstrated quantitative cell-based in vitro sensing and in vivo imaging of ligand-induced translocation of androgen receptor, which allowed high-throughput screening of exogenous and endogenous agonists and antagonists of this receptor.
The same authors used a similar approach to noninvasive molecular imaging of physical and emotional stress by developing a method for detecting physiological increases in the endogenous corticosterone caused by exo- and endogenous stress in living animals [46]. They constructed a pair of genetically encoded indicators composed of cDNAs of glucocorticoid receptor (GR), split Rluc, and a DnaE intein. The GR fused with C-terminal halves of Rluc and DnaE was localized in the cytosol, whereas a fusion protein of N-terminal halves of Rluc and DnaE was localized in the nucleus. If corticosterone induces GR translocation into the nucleus, the C-terminal Rluc meets the N-terminal one in the nucleus, and full-length Rluc is reconstituted by protein splicing with DnaE. Cell-based methods provided a quantitative bioluminescence assay of the extent of GR translocation into the nucleus. The authors further demonstrated that the indicator enabled noninvasive imaging in mice subjected to two different types of imposed stress: a forced swimming and metabolic perturbation caused by 2-deoxy-d-glucose. This stress indicator should be valuable for screening pharmacological compounds and in studying mechanisms of physiological stress.
Kanno et al. [47] also developed a genetically encoded bioluminescence indicator for monitoring the release of proteins from the mitochondria in living cells. The principle of this method is based on reconstitution of split Rluc fragments by protein splicing with a DnaE intein. A target mitochondrial protein connected with an N-terminal fragment of Rluc and an N-terminal fragment of DnaE is expressed in mammalian cells. If the target protein is released from the mitochondria toward the cytosol upon stimulation with a specific chemical, the N-terminal Rluc meets the C-terminal Rluc connected with C-terminal DnaE in the cytosol, and thereby, the full-length Rluc is reconstituted by protein splicing. The extent of release of the target fusion protein was evaluated by measuring activities of the reconstituted Rluc. To test the feasibility of this method, the authors monitored the release of Smac/DIABLO protein from mitochondria during apoptosis in living cells and mice. Their method allowed high-throughput screening of an apoptosis-inducing reagent, staurosporine, and imaging of the Smac/DIABLO release in cells and in living mice. This rapid analysis may be used for screening and assaying chemicals that would increase or inhibit the release of mitochondrial proteins in living cells and animals.
The split-intein system generally suffers from slow kinetic response rates posing problems for quantitative interrogation of reversible biochemical reactions, drug-induced protein associations, or shifts in equilibrium states of interacting proteins. The truly important aspects of studying PPIs in living cells or animals lie in the ability to do so in real time. The inevitable delay in the ability to detect an interaction using the split-intein strategy can be attributed to the time required for the splicing reaction. While this may not be a factor for slow reactions occurring over long time frames, numerous drugs, chemicals, and natural ligands exert their effects in seconds to minutes. The system also exhibits a high false-positive rate on account of the split-intein fragments being so small that at times it is believed they act merely as simple linker proteins, thus limiting the quantification of protein interactions.
9.4.2 Protein-Fragment Complementation Assays Using Split Firefly Luciferase
The most commonly used bioluminescence reporter gene for research purposes has been the luciferase from the North American Firefly (Photinus pyralis; Fluc). Fluc (61 kDa) catalyzes the transformation of its substrate D-Luciferin into oxyluciferin in a process dependent on ATP, Mg++, and O2, leading to self-emission of light from green to yellow wavelengths (560–610 nm, peak emission at 562 nm). In 2002, we demonstrated for the first time the feasibility of imaging PPIs using split reporters in small living animals [ 31 ]. We studied a PCA based on split Fluc (cleaved into two fragments, nFluc: residues 1–437; and cFluc: residues 438–550), using the interaction of Id and MyoD as test proteins (Fig. 9.1).
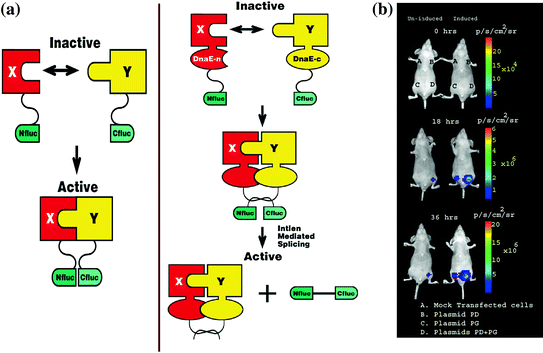
Fig. 9.1




Schematic diagram of the split reporter–based complementation strategy used to optically image PPIs in living mice. The N-terminal half of firefly luciferase is attached to protein X through a short peptide linker FFAGYC, and the C-terminal half firefly luciferase is connected to protein Y through the peptide linker CLKS. Interaction of proteins X and Y recovers Fluc activity through protein complementation (Fig. 9.1a). In vivo optical cooled CCD camera imaging of mice carrying transiently cotransfected 293T cells for the induction of the complementation-based split-luciferase system. All images shown are the visible light image superimposed on the optical CCD bioluminescence image with a scale in p/sec/cm2/sr. Mice were imaged in a supine position after IP injection of D-Luciferin. (Fig. 9.1b). A set of nude mice was repetitively imaged after subcutaneous implantation of 293T cells transiently cotransfected with various plasmids as described in Paulmurugan et al. [31]. One group of mice was induced with TNF-α, and the other group was not induced. The images were one representative mouse from each group immediately after implanting cells (0 h), and 18 and 36 h after TNF-α induction. The induced mouse showed higher Fluc signal at site D (where interacting proteins result in reporter protein complementation) when compared with the mouse not receiving TNF-α. The Fluc signal significantly increases after receiving TNF-α. Reproduced with permission from Paulmurugan et al. [31]
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
