82
CHAPTER OUTLINE
The endocrine effects of alcohol and other drugs are complex. These substances can alter the secretion of hormone-releasing factors and hormones at the level of the hypothalamus and pituitary; alter the synthesis or release of hormones at the level of the thyroid, adrenal, or pancreas; alter hormone action on target organs; and alter hormone economy by affecting their metabolism or binding proteins. Multiple hormone systems can be affected; many of whose actions may conflict, leading to a lack of net clinical effect. When one adds to this inherent complexity the effects on the endocrine system of gender, mental status, and mental illness, the heterogeneity of illicit drugs, the prevalence of polydrug use, the effects of drug withdrawal, and differences between the effects of acute and chronic substance use, it becomes clear that the study of the endocrine effects of alcohol and other drugs is very difficult. Generalizing many of these effects to clinical practice is even more difficult. Therefore, the most clearly recognized and clinically relevant effects are summarized in this chapter (Table 82-1).
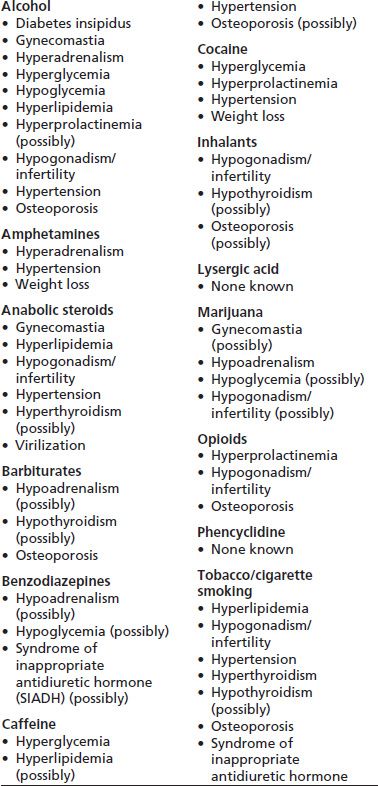
TABLE 82-1 ENDOCRINE SYNDROMES ASSOCIATED WITH ALCOHOL AND OTHER DRUG USE
DISORDERS RELATED TO ALCOHOL
Hypoglycemia
One of the most serious consequences of heavy alcohol use is hypoglycemia, which can lead to neurologic damage, coma, seizures, or death. Alcohol causes hypoglycemia by producing malnutrition, reducing the body’s production of glucose, and impairing the body’s response to hypoglycemia. Alcohol use is a common cause of spontaneous hypoglycemia (1,2).
Unlike other drugs, ethyl alcohol is a significant source of energy, with 7.1 kcal/g. Malnutrition can result from heavy alcohol use, as nutrients are replaced by the “empty” calories of alcohol (i.e., alcohol provides energy but no other nutrients such as protein or vitamins). In addition, the normal metabolism of alcohol to acetaldehyde by alcohol dehydrogenase leads to the conversion of nicotinamide adenine dinucleotide (NAD) to its reduced form (NADH), resulting in a decrease in gluconeogenesis. NAD is a cofactor necessary for many of the reactions involved in glucose synthesis. Alcohol does not affect glucose release from glycogen stores, which in a well-fed subject can provide glucose for 12 hours or more. In a prolonged fasting state or in a state of depleted glycogen stores, as seen in people with alcohol use disorders who are malnourished alcoholics, the inability to compensate for hepatic glycogen depletion can lead to hypoglycemia after acute alcohol ingestion.
Alcohol also can impair the body’s hormonal responses to hypoglycemia. Several investigators have shown that alcohol impairs the release of cortisol, growth hormone, glucagon, and vasopressin in response to hypoglycemia (3–7). Kolaczynski et al. (8) also found similar alcohol-induced reductions in cortisol, glucagon, and growth hormone levels in response to hypoglycemia but found faster glucose recovery with alcohol use, suggesting alcohol-related peripheral insulin resistance. Kerr et al. (9) found a similar suppression of growth hormone, an insignificant suppression of cortisol, and increased insulin resistance in mildly intoxicated hypoglycemic type 1 diabetics. Rasmussen et al. (10) found no change, on the other hand, in glucose counterregulation during acute insulin-induced hypoglycemia in diet-treated patients with mild type 2 diabetes.
There are five classes of alcohol-induced hypoglycemia: simple alcohol-induced fasting hypoglycemia, alcoholic ketoacidosis with hypoglycemia, alcoholic exacerbation of insulin-induced hypoglycemia, alcoholic exacerbation of sulfonylurea-induced hypoglycemia, and alcohol-induced reactive hypoglycemia. In simple alcohol-induced fasting hypoglycemia, patients typically present with coma and a blood glucose level of less than 40 mg/dL. Blood alcohol levels may be detectable. Because of the mechanism underlying this type of hypoglycemia, patients respond promptly to intravenous dextrose but may not respond to glucagon administration.
Alcoholic ketoacidosis is common. Depletion of NAD resulting from alcohol metabolism leads to the increased production of beta-hydroxybutyrate, acetoacetate, and acetone. Beta-hydroxybutyrate, which is not detectable by routine urine or serum ketone testing, predominates, so the beta-hydroxybutyrate-to-acetoacetate ratio can be as high as 7:1 to 10:1, as compared to 3:1 in diabetic ketoacidosis. Insulin secretion, which ordinarily prevents the development of ketoacidosis, is suppressed by decreased serum glucose levels and high catecholamine levels. The patient usually has a history of chronic heavy alcohol use and may be experiencing nausea, vomiting, abdominal pain, and metabolic acidosis. Blood alcohol levels usually are not detectable. Glucose levels may range from low to mildly elevated but typically are less than 250 mg/dL. Treatment includes dextrose and volume repletion. Marked hyperglycemia should raise the suspicion of concomitant diabetes or diabetic ketoacidosis (11).
Patients with diabetes treated with insulin or sulfonyl-urea are at risk for severe hypoglycemia with alcohol use (12–14). Cognitive impairment and nonrecognition of hypoglycemia with alcohol use can lead to nontreatment of hypoglycemia in diabetic patients (15,16). Patients with diabetes must be counseled about alcohol use and its hypoglycemic consequences.
Insulin and sulfonylureas should be used with great caution in patients with alcohol use disorders and diabetes, though they often are the only reasonable options for achieving some measure of glucose control in patients who continue to drink heavily. In addition, sulfonylureas and insulin are metabolized in the liver and are more likely to cause severe hypoglycemia in patients with alcoholic cirrhosis than in those without cirrhosis. Agents such as metformin, rosiglitazone, and pioglitazone, which lower glucose but do not produce hypoglycemia, may not be used because of the increased risk of lactic acidosis (metformin) and liver toxicity (rosiglitazone and pioglitazone). Short-acting insulin secretagogues of the meglitinide class, such as repaglinide and nateglinide, have been touted as causing less hypoglycemia and might be considered as having a role in patients with alcohol use disorders. However, Bolen et al. (17) have reported that hypoglycemia with repaglinide was comparable to that seen with sulfonylureas. Comparison of nateglinide with repaglinide has suggested a lower, albeit statistically insignificant, incidence of hypoglycemia (18). Cases of severe hypoglycemia have been described with repaglinide (19) and nateglinide (20).
Newer therapies such as glucagon-like peptide 1 agonists, such as exenatide and liraglutide, and dipeptidyl peptidase IV inhibitors, such as sitagliptin, saxagliptin, and linagliptin, increase incretin action, leading to glucose-dependent insulin release, reduced glucagon, and slowed gastric emptying. When used as monotherapy, they are more likely to cause hypoglycemia than placebo but less likely than sulfonylureas (21). These therapies have been associated with pancreatitis and gastrointestinal symptoms and may not be ideal for all patients with alcohol use disorders. Other therapies have been approved for improving glycemic control in type 2 diabetes mellitus, such as colesevelam, a bile acid resin, and bromocriptine, a dopamine agonist, and may be adjunctive therapies, but the former is not indicated in those with hypertriglyceridemia, and the latter may increase the risk for orthostatic hypotension and psychosis.
Alcohol abstinence is recommended, as is consumption of food with alcohol, if alcohol use persists, to minimize the risk of hypoglycemia. The American Diabetes Association recommends limiting alcohol intake (women: ≤1 drink daily and men: ≤2 drinks daily), consumption of food with alcohol to reduce the risk of nocturnal hypoglycemia, and minimizing use of mixers with significant carbohydrate content (22). Diabetes care in those with alcohol-related liver disease and ongoing heavy alcohol use is difficult, and less stringent glycemic goals as well as early consultation with a diabetologist are reasonable.
Reactive or postprandial hypoglycemia with alcohol use can occur, depending on the carbohydrate composition of the meal consumed. O’Keefe and Marks (23) first described this phenomenon as profound hypoglycemia (mean blood glucose nadir, 48.6 mg/dL) that occurs 3 to 4 hours after the ingestion of alcohol combined with 60 g sucrose (as in gin and tonic). This phenomenon can develop in up to 10% to 20% of healthy subjects and does not appear to occur with saccharin or fructose and alcohol (24). The quinine content of regular tonic, which increases insulin levels, was postulated as the cause of the hypoglycemia, but work by Flanagan et al. (25) suggests that the hypoglycemia develops because of suppression of growth hormone release.
Hyperglycemia
Alcoholic pancreatitis can lead to pancreatic exocrine and endocrine insufficiency. When diabetes mellitus results from pancreatic insufficiency, it is an indication that more than 90% of the pancreatic beta cells, which produce insulin, and the alpha cells, which produce glucagon, are destroyed. The secondary diabetes mellitus that results typically is an extremely labile insulin-dependent diabetes, in that patients are absolutely insulin deficient and have normal or increased insulin sensitivity. Diabetic ketoacidosis results not simply from a lack of insulin but from an increase in the glucagon–insulin ratio. This explains why type 2 diabetics may develop ketoacidosis in the setting of severe infection and the presence of high insulin levels. In patients with pancreatitis-induced diabetes, the lack of glucagon makes the patient somewhat resistant to developing diabetic keto-acidosis but also quite prone to developing hypoglycemia.
Acute ethanol consumption has been shown to increase peripheral insulin resistance, and interested readers are directed to an extensive review from Ting and Lautt (26). This insulin resistance is localized to skeletal muscle and may be related to ethanol-induced changes in lactate, triglyceride, norepinephrine, hepatic insulin–sensitizing substance, hepatic glutathione, and insulin binding. Despite this insulin resistance, postprandial glucose levels may be lower after preprandial consumption of 20 g of alcohol in lean, young healthy adults (27) and 15 g of alcohol after low-carbohydrate, high-fat meals in postmenopausal women (28).
Though chronic heavy alcohol use can lead to chronic pancreatitis, pancreatic insufficiency, and subsequent diabetes mellitus, some studies have found an association between low amounts alcohol use and improved insulin sensitivity (29–31) and a decreased risk of diabetes mellitus in men (32). Other investigators found no effect (33,34). The association of alcohol use and glucose control is a complicated one, with most studies finding a U-shaped association between alcohol use and diabetes mellitus, with low consumption having the lowest risk for diabetes mellitus (35,36). Some authors have found no association (37). There may be a threshold dose and duration effect as Davies et al. (38) found 30 g daily for 8 weeks was necessary to show decreases in insulin resistance. Daily doses of alcohol greater than 48 g may lead to a loss of this insulin sensitivity (39), resulting in a U-shaped relationship. Increases in adiponectin have been suggested as a cause for this decrease in insulin resistance (40). Ting and Lautt (26) have suggested increases in hepatic glutathione and hepatic insulin–sensitizing substance as a possible etiology for chronic alcohol’s effects on insulin resistance. The effects on alcohol on diabetes risk and control have recently been reviewed (41).
Reproductive Consequences
The reproductive effects of alcohol use are gender specific. Alcohol use long has been known to cause sexual dysfunction and hypogonadism in men, particularly in those with alcoholic cirrhosis (42). Even in men without liver disease, alcohol use leads to a decrease in testosterone (43), which may be the result of a combination of direct effects on the testicular synthesis of testosterone and hypothalamic– pituitary function (44) or on increases in sex hormone–binding globulin, which can lead to a decrease in bioavailable testosterone (45). The incidence of gynecomastia is increased in alcoholic cirrhosis, primarily because of increased levels of androstenedione, a precursor for estrogen synthesis (46). Increased prolactin levels have been found in patients with alcoholic cirrhosis (47), though it may not be present in those with alcohol use disorders who do not have severe liver disease (48).
In women, the effects of alcohol use depend on menopausal status and hormone therapy use. In premenopausal women and postmenopausal women on estrogen, acute alcohol consumption leads to an increase in estradiol levels through reduced metabolism of estradiol (49). This increase in estrogen level may explain why alcohol use is associated with an increased risk of breast cancer (50) and why alcohol can be associated with a delay in menopause. Women with high or frequent alcohol intake were found to have higher rates of menstrual disorders, including amenorrhea, dysmenorrhea, and irregular menstrual periods. Pregnant women with a high alcohol intake have a higher incidence of miscarriages, placental abruption, preterm deliveries, and stillbirths than do controls (51). An increased risk of infertility was associated with as few as one to five alcoholic drinks a week in a Danish study (52) and with as little as 12 g of alcohol a week (about one drink) in an American study (53). There may be an increase in prolactin levels and decrease in oxytocin levels in lactating women (54) and pre-menopausal women (55). Despite the increase in prolactin levels, lactating women may have a slight decrease in milk production (56).
Bone Health Consequences
Moderate to severe alcohol use disorders are associated with decreased bone mass and an increased risk of skeletal fractures (57–60). African American men may be less susceptible to this bone loss (61). Postmenopausal women who drink low amounts of alcohol may have an increased bone mineral density and a decreased risk of fractures (51). Similar effects have been found by others (62–64) with some finding differences in the type of alcoholic beverage, with wine or beer being associated with more benefit than spirits or liquor (65,66).
In addition to alterations in sex hormones and the type of fat malabsorption and vitamin D deficiency associated with liver disease, alcohol use can reversibly impair bone formation by osteoblasts (67–69). Malnutrition and low body mass and decreases in IGF-I, a bone growth factor, may play a significant role in the decreased bone mass (70,71). Reductions in leptin with alcohol use (72,73) may also play a role in decreasing bone mass although there is some controversy about exactly what that role is (74). In animal studies, parathyroid hormone may help reverse some of the bone loss though continued alcohol use impairs the response (75). The effects of alcohol on bone were recently extensively reviewed (76).
Heavy alcohol use is associated with osteonecrosis of bone (77). Alcohol-associated osteonecrosis of the femoral head may be due in part to lipid or cortisol changes (78) and, in fact, alcohol may be responsible for up to one-third of cases of femoral head osteonecrosis (79).
Other Endocrinologic Consequences
Alcohol use increases triglyceride synthesis, leading to hypertriglyceridemia and hepatic steatosis. The increased NADH/NAD ratio leads to increases in alpha-glycerophos-phate, which favors hepatic triglyceride accumulation by trapping fatty acids. The excess NADH enhances fatty acid synthesis. Alcohol increases the high-density lipoprotein (HDL) fraction of cholesterol, which may be associated with reduced cardiovascular morbidity and mortality (80).
Though glucocorticoid response to hypoglycemia can be impaired by alcohol use, patients with chronic heavy alcohol use have elevated adrenocorticotropic hormone (ACTH) and cortisol production. This response is mediated by corticotro-pin-releasing hormone and the interaction with stress. This may be related to a person’s genetic propensity to developing an alcohol use disorder. These effects are dose dependent (81).
Rarely, these patients may develop the clinical stigmata of glucocorticoid excess, such as central obesity, moon facies, “buffalo hump,” and biochemical evidence of nonsuppressible glucocorticoid excess that is difficult to distinguish from Cushing syndrome. This condition, which has been termed pseudo-Cushing syndrome, reverses with abstinence from alcohol for at least 1 month (82,83). There are no clear clinical features or lab test results that distinguish pseudo-Cushing’s from Cushing’s (83), and a period of alcohol abstinence and reversal of the syndrome may avoid the search for a nonexistent pituitary or adrenal tumor.
Alcohol may influence hormones regulating feeding behavior. Leptin levels are decreased acutely after low amounts of alcohol use (72) and in malnourished people with an alcohol use disorder (84). Ghrelin secretion is similarly reduced, without effects on peptide YY, soon after moderate alcohol intake (85,86). This is somewhat para-doxic given these hormones suppress appetite, and alcohol is believed to stimulate appetite.
Alcohol use has effects on body volume and blood pressure status. It transiently decreases vasopressin release, leading to water diuresis (87). This condition can be problematic in patients with partial diabetes insipidus. Acute alcohol use also increases blood pressure, possibly through an increase in norepinephrine (88). Low amounts of alcohol consumption appears to increase plasma renin activity, though such increases are believed to result from a secondary response to changes in fluid and electrolyte balance (89). Though alcohol by itself does not affect mineralocorticoid levels, alcoholic cirrhosis leads to elevations of aldosterone in response to decreased effective plasma volume.
Acute alcohol use does not produce thyroid dysfunction in patients with previously normal thyroid function (90). Alcoholic cirrhosis of the liver can produce decreases in serum triiodothyronine (T3) because the liver is a major site for the conversion of thyroxine to T3, without producing clinical hypothyroidism in those without preexisting autoimmune thyroid disease (91,92).
Alcohol use was reported to suppress melatonin secretion through increased norepinephrine levels, and this increase may be implicated in disturbances in sleep and performance (93).
DISORDERS RELATED TO TOBACCO
The adverse effects of tobacco on the cardiopulmonary systems are well known. Tobacco smoke contains a myriad of chemical compounds—most notably nicotine; tar; thiocya-nate; 2,3-hydroxypyridine; and carbon monoxide—which may have multiple endocrine effects. Many of these effects have been recently reviewed in detail (94).
Thyroid Disease
Cigarette smoking increases the risk of Graves disease and Graves ophthalmopathy and can lower thyroid-stimulating hormone levels (95–101). Smoking cessation seems to be associated with reduction in the risk for Graves disease (102). Passive smoke exposure may increase resting metabolic rate and thyroid hormone secretion (103,104). The increased risk of ophthalmopathy with cigarette smoking may be related to increased fibroblast adipogenesis associated with increased interleukin-1 levels (105).
Smoking may also be associated with decreased levels of serum thyroid autoantibodies (99,106–109), with smoking cessation increasing the risk for these antibodies (110). Smoking appears to protect smokers from hypothyroidism (99,108) and goiter (111,112). However, older studies have associated smoking with goiter and hypothyroidism (113,114). This may be a consequence of the thiocyanate present in cigarette smoke, which is a goitrogen that inhibits iodide uptake and hormone synthesis and increases iodine exit from the thyroid (115,116). Smoking may cause hypothyroidism only in those with preexisting thyroid dysfunction, and there may be impaired peripheral action of thyroid hormone (117). Smoking appears to lower the risk for thyroid cancer (118,119).
Insulin Resistance and Dyslipidemia
Cigarette smoking (120), as well as nicotine gum use (121), is associated with an increase in insulin resistance and an increased risk of developing impaired glucose tolerance and diabetes mellitus (32,122–125). Secondhand smoke exposure similarly increases diabetes risk (126). Mild decreases in HDL cholesterol and mild elevations in triglycerides, consistent with this insulin resistance, are associated with cigarette smoking (120), though these changes may be due to components of cigarette smoke other than nicotine (127). Both passive smoke exposure and active smoking have been associated with the metabolic syndrome in adolescents (128). Insulin resistance and dyslipidemia may be responsible, in part, for the elevated rates of cardiovascular and atherosclerotic disease associated with cigarette use.
Reproductive Function
In women, cigarette smoking is associated with decreases in estrogen levels (129) and, in fact, can enhance estrogen degradation (130). It also is associated with early menopause (131,132) and increased ovarian age and follicular-stimulating hormone (FSH) levels (133,134). A recent meta-analysis found that smoking patients had poorer outcomes with assisted reproduction, with significantly lower odds of clinical pregnancy per cycle, higher odds of spontaneous miscarriage, and higher odds of ectopic pregnancy (135). In men, cigarette smoking is associated with quantitative and qualitative decrements in sperm (136–138) and with increases in serum estrogen (139). Unfortunately, public knowledge of the risk of smoking for these reproductive issues is low, ranging from 17% to 39% (140), and educating patients about these unrealized dangers is paramount.
Bone Health
Cigarette smoking is associated with decreased bone mineral density and increased bone loss in postmenopausal women and elderly men (141) and is an independent risk factor for osteoporotic fracture (142). This topic was recently reviewed (143). This fracture risk appears, in part, due to a decrease in intestinal calcium absorption (144,145), resulting in a negative calcium balance. Brot et al. (146) found an association between smoking and decreased serum 25- hydroxyvitamin D, 1,25-dihydroxyvitamin D, parathyroid hormone, and osteocalcin levels, suggesting a more complex effect of smoking on calcium and bone metabolism. Cigarette smoking can negate the protective effect of estrogen therapy on the risk of hip fracture in postmenopausal women (147). It also is associated with femoral head osteonecrosis (77), possibly through impairment of vascular function or changes in blood lipids.
Other Endocrinologic Effects
Cigarette smoke stimulates antidiuretic hormone release from the pituitary through an airway-mediated mechanism that does not depend on circulating nicotine (148,149). This antidiuretic effect may cause or exacerbate hyponatre-mia in susceptible patients (150,151). The nicotine found in cigarette smoke can increase the release of catecholamines from the adrenal medulla, which may precipitate a hypertensive crisis for those with pheochromocytoma. In addition, smoking is associated with hypertension and poorly controlled hypertension, apparently through stimulation of the noradrenergic nervous system (152); such hypertension is not angiotensin II–dependent (153). Nicotine increases ACTH release and cortisol release in a dose-dependent fashion (154–156) and may have some role in tobacco addiction. Cigarettes also may increase prolactin secretion (154,156).
DISORDERS RELATED TO OTHER DRUGS
Marijuana
The major psychoactive component of marijuana is delta-9-tetrahydrocannabinol (THC), though marijuana can contain more than 400 chemicals. Factors including concomitant use of other drugs, variability in smoking technique, and variable potencies of marijuana cigarettes and development of tolerance to marijuana’s effects can make it difficult to generalize from study findings to clinical practice. Consistent endocrine effects of marijuana in animals have not translated to consistent findings in humans (157).
Though some investigators have reported that heavy marijuana smoking is associated with low plasma levels of testosterone (158), others report that chronic marijuana use has no effects on plasma testosterone or luteinizing hormone (LH) in men (159). Further confusion arises from reports suggesting that smoking marijuana cigarettes acutely suppresses LH in men (160) and premenopausal women in the luteal phase of their menstrual cycle but not in postmenopausal women (161). One study of pregnant marijuana users found no changes in female sex hormones as compared with controls (162). The many conflicting studies make it unlikely that chronic marijuana use affects the reproductive system with any significant clinical relevance, and interested readers are referred to a recent extensive review (163). Marijuana has not been found to be teratogenic and has not been found to have consistent effects on pregnancy outcomes. There have been recent reports of an association with chronic marijuana use and increased risk of testicular germ cell tumors (164,165).
With regard to other endocrinologic effects, prolonged oral administration of 210 mg THC a day (the equivalent of smoking six marijuana cigarettes) for 14 days significantly suppressed the cortisol and growth hormone response to insulin-induced hypoglycemia, though the THC use did not suppress the cortisol and growth hormone response to values consistent with cortisol or growth hormone deficiency (166). Continued marijuana use has not been found to affect the normalization of the hypothalamic–pituitary–adrenal axis in heroin addicts initiating methadone treatment (167). Adolescents with reported early onset (9 to 12 years of age) of marijuana use were found to have lower salivary cortisol levels on awakening (168). It is unclear whether the lower salivary cortisol resulted from the early marijuana use or vice versa. Heavy marijuana use may likely cause significant hypoadrenalism or impaired recovery from insulin-induced hypoglycemia only in those with preexisting adrenal disease. In animals, marijuana suppresses TSH and peripheral thyroid hormone levels (157), and one recent study in humans showed a normal TSH but decreased peripheral T4 associated with an increased T3 uptake, suggesting dose-dependent decreases in thyroid-binding globulin (169), but no clear alteration in thyroid status. Marijuana has well-recognized effects on feeding behavior (“the munchies”) by its interactions with cannabinoid receptors (CB-1) (170). In a recent study in HIV-infected men, marijuana smoking increased serum leptin and ghrelin and decreased peptide YY levels, without affecting insulin levels (171).
Opioids
The endocrine effects of acute administration of opioids occur primarily in the hypothalamus and pituitary. Gonadotropins (FSH and LH) are suppressed by inhibition of gonadotropin-releasing hormone secretion. Prolactin secretion is stimulated, while ACTH and cortisol secretion is suppressed (172). Chronic administration of opioids can produce partial tolerance to many of the endocrine effects. Vuong et al. (173) have recently exhaustively reviewed the endocrine effects of opioids.
Male gonadal function in methadone and heroin addicts has been primarily diminished but not in all studies. In men, methadone use has been associated with a decline in serum testosterone levels in several early studies (174–176). Cushman and Kreek did not find changes in testosterone level among people with heroin addiction maintained on methadone maintenance (177,178). Another study looking at methadone, methadone/heroin, and heroin use found normal levels of testosterone, FSH, LH, and prolactin in all but the heroin users who had elevated prolactin levels (179). All the heroin-addicted, the heroin/methadone users, and 45% of the methadone takers in that study had diminished sperm motility. Lafisca et al. (180) looked at short-term methadone use in 30 men and found no abnormalities of estrogen, progesterone, or LH, whereas FSH showed lower values than normal, and androstenedione, dehy-droepiandrosterone, and prolactin noticeably increased in many subjects. Modest variations were noted for testosterone and dihydrotestosterone. Other investigators found a decrease in basal FSH and LH levels, with a decrease in pituitary response to gonadotropin-releasing hormone, suggesting a hypothalamic cause for hypogonadism in people with heroin addiction (181). While Brown et al. (182) found sexual dysfunction associated with methadone dose in 14% of men attending a methadone maintenance clinic, they did not find it associated with a decrease in testosterone level.
Daniell (183) found men being treated with sustained-action opioids for chronic pain had decreasing testosterone levels in a dose-dependent fashion, with overtly low levels with a daily methadone dose equivalent or greater than 70 mg. Many were overtly symptomatic with erectile dysfunction. Bliesener et al. (184) found that people with opioid addiction on buprenorphine maintenance had higher testosterone levels, comparable to normal healthy controls, than those on methadone maintenance suggesting that hypogonadism may be mediated through mu opioid receptor since methadone is a pure mu opioid receptor agonist and buprenorphine is a mu opioid receptor agonist with high affinity but low intrinsic activity. Hallinan et al. (185,186) confirmed that male hypogonadism and sexual dysfunction were more common with methadone maintenance therapy than with buprenorphine.
The variability in opioid-induced male hypogonadism may be due in part to the presence of other components, as well as variable doses of opioids, in street preparations of heroin, continued use of heroin in methadone-treated addicts, and the possible effect of malnutrition on the reproductive system. A study of the endocrine effects of long-term intrathecal opioid administration for pain relief found diminished testosterone and LH levels in men but normal FSH and prolactin levels in comparison to control patients with a comparable pain syndrome but who were not treated with opioid (187), suggesting that these are the “pure” opiate effects on the male reproductive system.
In women, more than half of a group of 76 former heroin addicts on methadone maintenance had menstrual irregularities (188). Endocrinologic studies on seven of the women showed alterations in hypothalamic and pituitary function leading to oligoovulation. A study of 21 premenopausal women who received chronic intrathecal opioids found 14 to have amenorrhea and 7 to have menstrual irregularity, with decrements in serum LH, FSH, estradiol, and progesterone concentrations (187). In the same study, 18 postmenopausal women had serum LH and FSH levels significantly lower than did postmenopausal control subjects, further indicating impairment of gonado-tropin release. Women being treated for chronic pain with sustained-action, transdermal, or oral opioids have been associated with lower sex hormones due to ovarian and adrenal suppression (189). Opioids have been found to be competitive inhibitors of human placental aromatase, the enzyme that converts androgens to estrogens (190,191). The clinical implications of this effect and generalizability to other tissues are unclear.
This hypogonadism may also be influenced through hyperprolactinemia. Opioids are well known to increase prolactin levels (192), and this is likely mediated through dopaminergic mechanisms (193). Naloxone does not affect TRH-stimulated release of prolactin (194).
Hypogonadism is a risk factor for osteoporosis. There is evidence showing a decrease in bone density with opioid (195–199) in both men and women, although two studies found it more common in men than in women and associated with a higher risk of hypogonadism (200,201). Kim et al. (202) found a high degree of vitamin D deficiency and insufficiency among those in a methadone maintenance program, which may adversely affect the bone. There is also evidence for increased fracture risk (203,204). The effects on bone may be reversed with cessation of the opioid (195).
Opioids have been associated with hypoadrenalism, whether given intrathecally (187), orally (205,206), or transdermally (207). Opioids appear to inhibit the ACTH response to corticotropin releasing hormone (CRH), and their effect is blocked by naloxone (208,209).
Opioids at high doses may induce growth hormone release among normal subjects but to a greater extent in those with acromegaly (210). Naloxone may block the stimulation of growth hormone with growth hormone– releasing hormone (211). Opioids may also stimulate TSH release (192), an effect blocked by naloxone (194).
Cocaine
Cocaine acts primarily by blocking the reuptake of norepi-nephrine, dopamine, and serotonin at the synaptic junctions, resulting in increased neurotransmitter concentrations. In animal studies, cocaine can stimulate the adrenal medulla to release epinephrine and norepinephrine. By increasing cat-echolamines, counterregulatory hormones that antagonize insulin, stimulate glucose production, and inhibit glucose clearance, hyperglycemia can result as can diabetic ketoacidosis or hyperosmolar nonketotic hyperglycemia without any other identified precipitants (212,213
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


