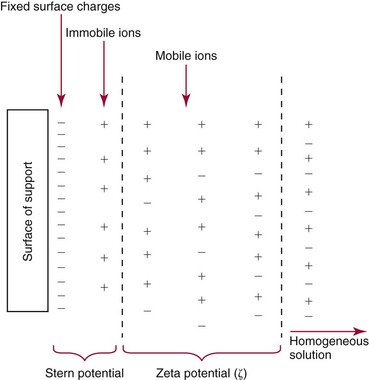
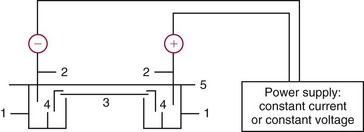
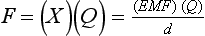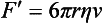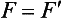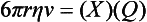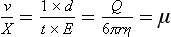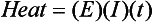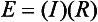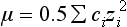Chapter 12 Depending on the charge they carry, ionized solutes move toward either the cathode (negative electrode) or the anode (positive electrode) in an electrophoresis system. For example, positive ions (cations) migrate to the cathode, and negative ions (anions) to the anode. An ampholyte (a molecule that is either positively or negatively charged, formerly called a zwitterion) becomes positively charged in a solution that is more acidic than its isoelectric point (pI)* and migrates toward the cathode. In a more alkaline solution, the ampholyte becomes negatively charged and migrates toward the anode. Because proteins contain many ionizable amino (—NH2) and carboxyl (—COOH) groups, and because the bases in nucleic acids may also be positively or negatively charged, they both behave as ampholytes in solution. The rate of migration of ions in an electrical field depends on the factors listed in Box 12-1. The equation expressing the driving force in such a system is given by where F = the force exerted on an ion X = the current field strength (V/cm) (i.e., voltage drop per unit length of medium) EMF = the electromotive force [voltage (V) applied] where r = the ionic radius of the solute η = the viscosity of the buffer solution in which migration is occurring v = the rate of migration of the solute = velocity, length (l) traveled per unit of time (cm/s) then or where v/X is the rate of migration (cm/s) per unit field strength (E/cm), defined as the electrophoretic mobility. It is expressed by the symbol µ and has the units cm2/(V)(s). In addition to the factors listed in Box 12-1, other factors that affect electrophoretic mobility include electroendosmosis (endosmosis) and wick flow. Electroendosmosis affects mobility by causing uneven movement of water through the support medium. An electrophoretic support medium, such as a gel in contact with water, takes on a negative charge caused by adsorption of hydroxyl ions. These ions are fixed to the surface and are immobile. Positive ions in solution cluster about the fixed negative charge sites, forming an ionic cloud of mostly positive ions. The number of negative ions in the solution increases with increasing distance from the fixed negative charge sites until eventually positive and negative ions are present in equal concentrations (Figure 12-1). Although modern electrophoresis equipment and systems vary considerably in form and degree of automation, the essential components common to all systems (Figure 12-2) include two reservoirs (1), which contain the buffer used in the process, a means of delivering current from a power supply via platinum or carbon electrodes (2), which contact the buffer, and a support medium (3) in which separation takes place connecting the two reservoirs. In some systems, wicks (4) may connect the medium to the buffer solution or directly to the electrodes. The entire apparatus is enclosed (5) to minimize evaporation and protect both the system and the operator. The direct current power supply sets the polarity of the electrodes and delivers current to the medium. Ionic strength (also denoted by the symbol µ) is computed according to the following: where A relatively high ionic strength buffer used in high-resolution electrophoresis improves the separation of serum proteins into as many as 13 bands, with two or more bands in the α1, α2, and β-globulin regions and one or more additional bands seen in various pathologic conditions. Because of higher conductivity and the associated heat produced, it is necessary to reduce the temperature of the system to 10 to 14 °C. “Submarine” techniques, in which gels are submersed in circulating buffer cooled by an external cooling device or are supported on an electrophoresis chamber cooled by circulating water or an integral Peltier plate,41 provide exact temperature control. Effective cooling with less precise temperature control may also be achieved using chambers designed with a sealed compartment of cooled ethylene glycol, which is in contact with the gel during running. Attempts to improve the hydrophilic nature of polyacrylamide have led to the development of mono- and di-substituted monomers, one of which is N-acryloyl-tris(hydroxymethyl)aminomethane, or poly NATTM.5 This material is more hydrophilic than polyacrylamide, and its matrix has larger pores, thereby presenting less resistance to the passage of large molecules. It is ideally suited to the separation of DNA fragments up to 20 kilobases (kb) in size using a homogeneous (nonpulsed) electric field. Fragments that differ in size by as little as 2% can be resolved. Gels are submersed in buffer during use, allowing temperatures to be tightly controlled at values between 50 and 60 °C. Use of elevated temperatures results in shorter run times and more reproducible band migration. Because of increased volume of testing, primarily for serum proteins, many laboratories are converting to automated systems for electrophoresis. Such a system is the Helena SPIFE 4000 (Helena Laboratories, Beaumont, Tex), an automated electrophoresis system providing automated reagent application and a variety of gel sizes that permit analysis of 10 to 100 samples simultaneously. It also features (1) in-line sample application, (2) automated electrophoretic separation and staining of analytes, (3) multiple staining capability, and (4) positive sample identification. The Interlab Microgel system (Interlab Srl, Rome, Italy) also fully automates the process and integrates (1) sample application, (2) temperature-controlled electrophoresis, (3) staining, and (4) densitometry into a single unit with the capability of managing four gels simultaneously. Other systems that have partially automated the procedure or incorporated the ability to process sequentially multiple gels of different compositions include the (1) Phast System (Pharmacia LKB, Gaithersburg, Md), (2) HITE Fractoscan (Olympus, Invicon, München, Germany), (3) Hydragel-Hydrasys (Sebia Inc., Durham, NC), and (4) High-Performance Gel Electrophoresis (HPGE)-1000 system (LabIntelligence Inc., Belmont, Calif). Most CE systems (see “Capillary Electrophoresis” section) have autosampling capability for sequentially processing specimens, but the Sebia Capillarys permits simultaneous processing of seven samples by using multiple capillaries. Newer microchip-based analyzers like the Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Santa Clara, Calif) significantly miniaturize and increase the speed of the process for separating proteins, nucleic acids, or even entire cells. These advances substantially reduce the labor component associated with this technique. Table 12-1 lists dyes commonly used in electrophoresis, along with suggested wavelengths for quantification by densitometry. Most commercial methods for serum protein electrophoresis use Amido Black B or members of the Coomassie Brilliant Blue series of dyes for staining. Isoenzymes are typically visualized by incubating the gel in contact with a solution of substrate, which is linked structurally or chemically to a dye before fixing. Silver nitrate and silver diammine stain proteins and polypeptides with sensitivity 10- to 100-fold greater than that of conventional dyes.52 Selective fixing and staining of protein subclasses also can be achieved by combining a stain molecule with an antiglobulin, as is done in immunofixation. TABLE 12-1 Suggested Wavelengths for Quantitation of Protein Zones by Direct Densitometry After Staining Improvements in conducting sensitive measurements have been achieved by linking an enzyme such as alkaline phosphatase or peroxidase to a single or double antibody specific for particular proteins such as oligoclonal immunoglobulin (Ig)G,53 or by spraying separated proteins with luminal and peroxide to develop chemiluminescence, which, in turn, exposes x-ray film to form a permanent image.64 Chemiluminescence has been used in this way to quantify IgE (Lumi-Phos 530, Lumigen, Inc., Southfield, Mich),67 and DNA fragments have been detected by linking with a fluorescent dye label.35 In 1975, Edward Southern developed a technique that is widely used to detect fragments of DNA. This technique, known as Southern blotting,24 first requires electrophoretic separation of DNA or DNA fragments by AGE. Next, a strip of nitrocellulose or a nylon membrane is laid over the agarose gel, and the DNA or DNA fragments are transferred or “blotted” onto it by capillary, electro-, or vacuum blotting. They are then detected and identified by hybridization with a labeled, complementary nucleic acid probe. This technique is widely used in (1) molecular biology for identifying a particular DNA sequence; (2) determining the presence, position, and number of copies of a gene in a genome; and (3) typing DNA (see Chapters 17, 38, and 39 for further details). Northern and Western blotting techniques,24 named by analogy to Southern blotting, were subsequently developed to separate and detect ribonucleic acids (RNAs) and proteins, respectively. Northern blotting is carried out identically to Southern blotting except that a labeled RNA probe is used for hybridization. Western blotting is used to separate, detect, and identify one or more proteins in a complex mixture. It involves first separating the individual proteins by polyacrylamide gel and then transferring or “blotting” onto an overlying strip of nitrocellulose or a nylon membrane by electro-blotting. The strip or membrane is then reacted with a reagent that contains an antibody raised against the protein of interest.
Electrophoresis
Theory Of Electrophoresis
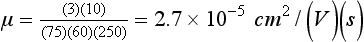
Conventional Electrophoresis
Instrumentation
Buffers
Support Media
Polyacrylamide Gel
Automated Systems
General Operations
Detection and Quantification
Staining

Blotting Techniques
Electrophoresis