32
Drugs of Abuse
CASE STUDY
Mr V, a 47-year-old man, was recently promoted as a director of a transportation company. A routine inspection of the books shows that a large sum of money is missing. Subsequent investigation finds that Mr V has been spending more than $20,000 a month to buy cocaine; currently he consumes 2–3 g/d. He also drinks several beers each day and 5–8 shots of vodka in the evening. He spends weekend nights in clubs, where he often consumes 2–3 pills of ecstasy. He began using drugs at age 18; during parties he mostly smoked cannabis (5–6 joints per weekend), but also tried cocaine. This “recreational use” came to an abrupt halt when he married at age 27 and entered a professional training program that allowed him to obtain his current job, now jeopardized by his cocaine use. Is Mr V addicted, dependent, or both? What is the reason for the use of several different addictive drugs at the same time?
Drugs are abused (used in ways that are not medically approved) because they cause strong feelings of euphoria or alter perception. However, repetitive exposure induces widespread adaptive changes in the brain. As a consequence, drug use may become compulsive—the hallmark of addiction.
 BASIC NEUROBIOLOGY OF DRUG ABUSE
BASIC NEUROBIOLOGY OF DRUG ABUSE
DEPENDENCE VERSUS ADDICTION
Recent neurobiologic research has led to the conceptual and mechanistic separation of “dependence” and “addiction.” The older term “physical dependence” is now denoted as dependence, whereas “psychological dependence” is more simply called addiction.
Every addictive drug causes its own characteristic spectrum of acute effects, but all have in common that they induce strong feelings of euphoria and reward. With repetitive exposure, addictive drugs induce adaptive changes such as tolerance (ie, escalation of dose to maintain effect). Once the abused drug is no longer available, signs of withdrawal become apparent. A combination of such signs, referred to as the withdrawal syndrome, defines dependence. Dependence is not always a correlate of drug abuse—it can also occur with many classes of nonpsychoactive drugs, eg, sympathomimetic vasoconstrictors and bronchodilators, and organic nitrate vasodilators. Addiction, on the other hand, consists of compulsive, relapsing drug use despite negative consequences, at times triggered by cravings that occur in response to contextual cues (see Box: Animal Models in Addiction Research). Although dependence invariably occurs with chronic exposure, only a small percentage of subjects develop a habit, lose control, and become addicted. For example, very few patients who receive opioids as analgesics desire the drug after withdrawal. And only one person out of six becomes addicted within 10 years of first use of cocaine. Conversely, relapse is very common in addicts after a successful withdrawal when, by definition, they are no longer dependent.
ADDICTIVE DRUGS INCREASE THE LEVEL OF DOPAMINE: REINFORCEMENT
To understand the long-term changes induced by drugs of abuse, their initial molecular and cellular targets must be identified. A combination of approaches in animals and humans, including functional imaging, has revealed the mesolimbic dopamine system as the prime target of addictive drugs. This system originates in the ventral tegmental area (VTA), a tiny structure at the tip of the brainstem, which projects to the nucleus accumbens, the amygdala, the hippocampus, and the prefrontal cortex (Figure 32–1). Most projection neurons of the VTA are dopamine-producing neurons. When the dopamine neurons of the VTA begin to fire in bursts, large quantities of dopamine are released in the nucleus accumbens and the prefrontal cortex. Early animal studies pairing electrical stimulation of the VTA with operant responses (eg, lever pressing) that result in strong reinforcement established the central role of the mesolimbic dopamine system in reward processing. Direct application of drugs into the VTA also acts as a strong reinforcer, and systemic administration of drugs of abuse causes release of dopamine. Even selective activation of dopamine neurons is sufficient to elicit behavioral changes typically observed with addictive drugs. These very selective interventions use optogenetic methods. Blue light is delivered in a freely moving mouse through light guides to activate channelrhodopsin, a light-gated cation channel that is artificially expressed in dopamine neurons. As a result, mice will self-administer blue light; pairing light activation of VTA dopamine neurons with a specific environment establishes a long-lasting place preference. Conversely using inhibitory optogenetic effectors or activation of inhibitory neurons upstream causes aversion.
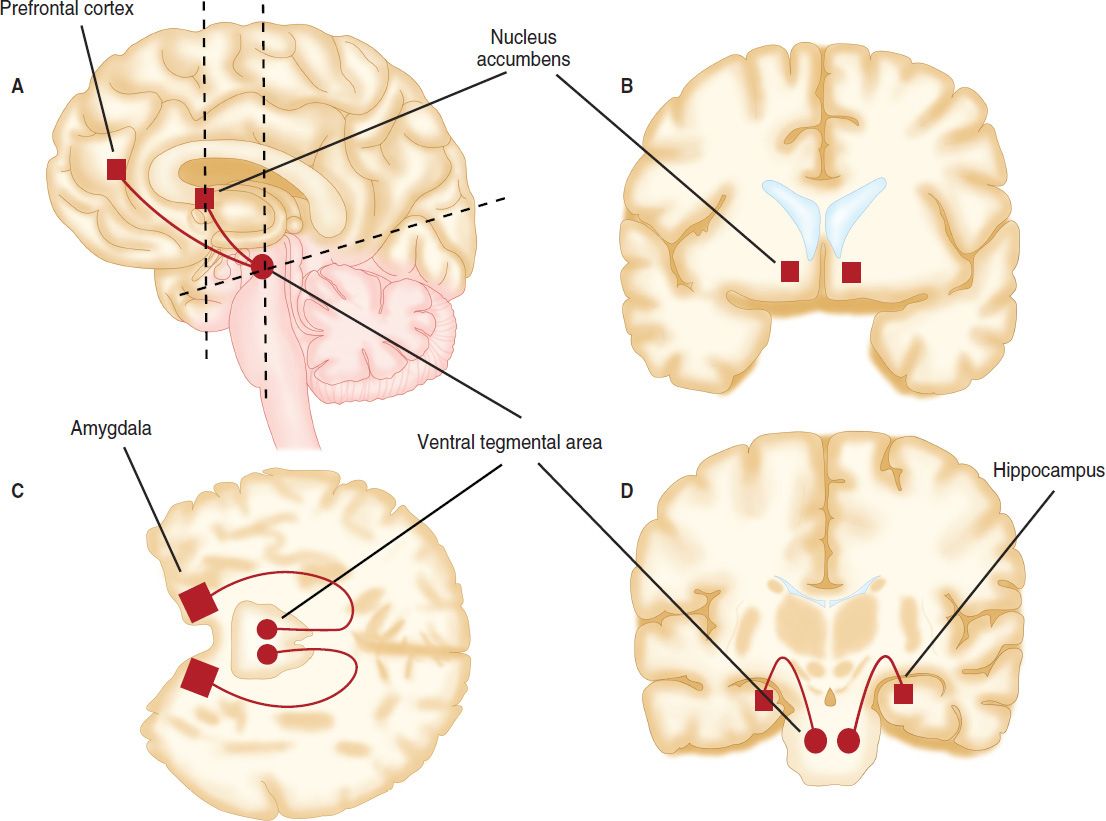
FIGURE 32–1 Major connections of the mesolimbic dopamine system in the brain. Schematic diagram of brain sections illustrating that the dopamine projections originate in the ventral tegmental area and target the nucleus accumbens, prefrontal cortex, amygdala, and hippocampus. The dashed lines on the sagittal section indicate where the horizontal and coronal sections were made.
As a general rule, all addictive drugs activate the mesolimbic dopamine system. The behavioral significance of this increase of dopamine is still debated. An appealing hypothesis is that mesolimbic dopamine codes for the difference between expected and actual reward and thus constitutes a strong learning signal (see Box: The Dopamine Hypothesis of Addiction).
Since each addictive drug has a specific molecular target that engages distinct cellular mechanisms to activate the mesolimbic system, three classes can be distinguished: A first group binds to Gio protein-coupled receptors, a second group interacts with ionotropic receptors or ion channels, and a third group targets the dopamine transporter (Table 32–1 and Figure 32–2). G protein-coupled receptors (GPCRs) of the Gio family inhibit neurons through postsynaptic hyperpolarization and presynaptic regulation of transmitter release. In the VTA, the action of these drugs is preferentially on the γ-aminobutyric acid (GABA) neurons that act as local inhibitory interneurons. Addictive drugs that bind to ionotropic receptors and ion channels can have combined effects on dopamine neurons and GABA neurons, eventually leading to enhanced release of dopamine. Finally, addictive drugs that interfere with monoamine transporters block reuptake or stimulate nonvesicular release of dopamine, causing an accumulation of extracellular dopamine in target structures. Since neurons of the VTA also express somatodendritic transporters, which normally clear dopamine released by the dendrites, class 3 drugs also increase dopamine level in the VTA. Although drugs of this class also affect transporters of other monoamines (norepinephrine, serotonin), action on the dopamine transporter remains central for addiction. This is consistent with the observations that antidepressants that block serotonin and norepinephrine uptake, but not dopamine uptake, do not cause addiction even after prolonged use.
TABLE 32–1 The mechanistic classification of drugs of abuse.1
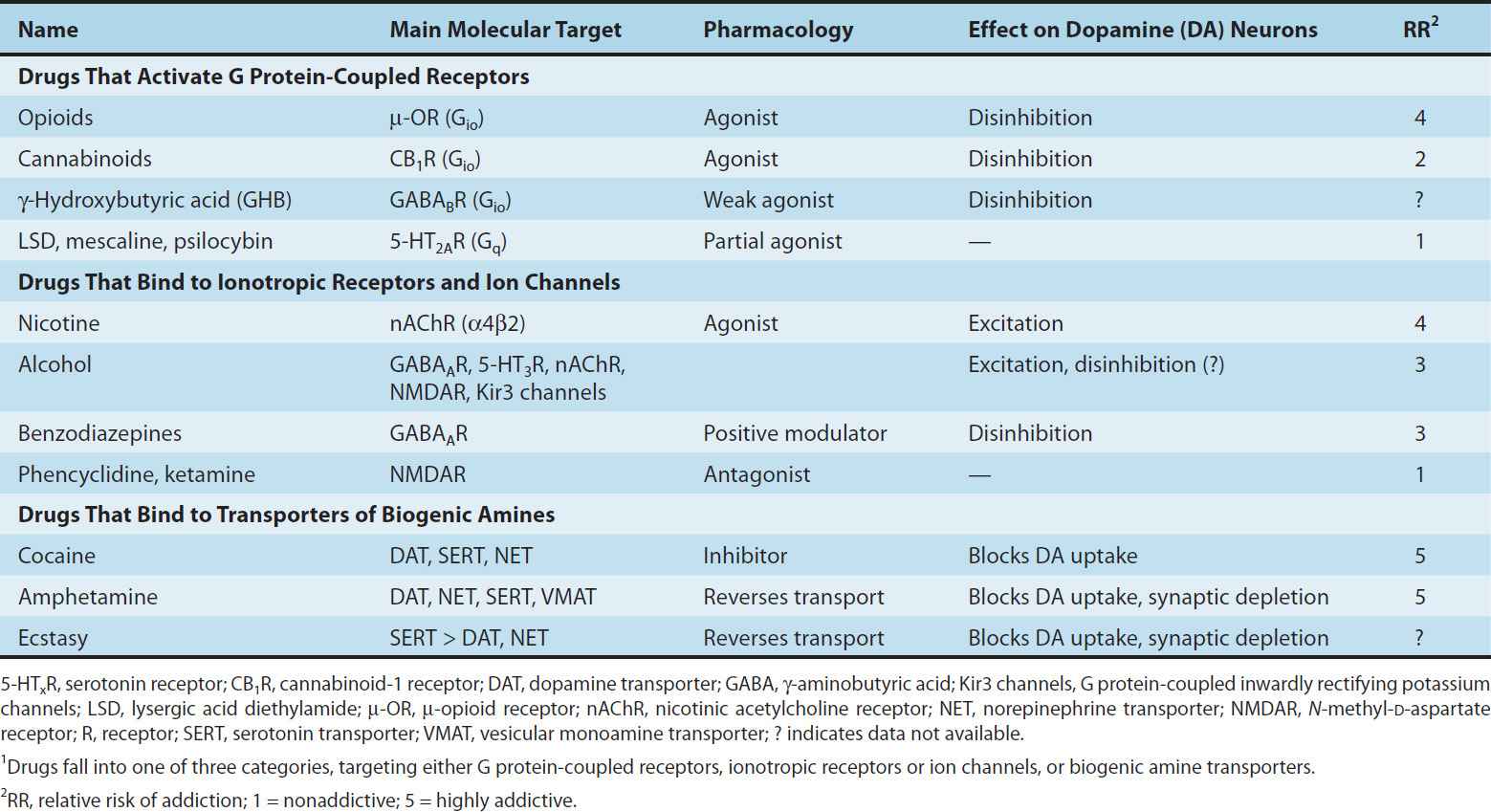
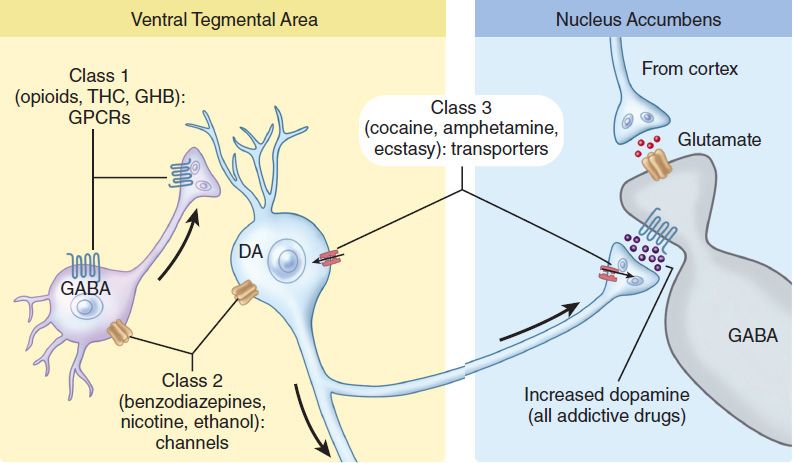
FIGURE 32–2 Neuropharmacologic classification of addictive drugs by primary target (see text and Table 32–1). DA, dopamine; GABA, γ-aminobutyric acid; GHB, γ-hydroxybutyric acid; GPCRs, G protein-coupled receptors; THC, Δ9-tetrahydrocannabinol.
Animal Models in Addiction Research
Many of the recent advances in addiction research have been made possible by the use of animal models. Since drugs of abuse are not only rewarding but also reinforcing, an animal will learn a behavior (eg, press a lever) when paired with drug administration. In such a self-administration paradigm, the number of times an animal is willing to press the lever in order to obtain a single dose reflects the strength of reinforcement and is therefore a measure of the rewarding properties of a drug. Observing withdrawal signs specific for rodents (eg, escape jumps or “wet-dog” shakes after abrupt termination of chronic morphine administration) allows the quantification of dependence. Behavioral tests for addiction in the rodent have proven difficult to develop, and so far no test fully captures the complexity of the disease. However, it is possible to model core components of addiction; for example, by monitoring behavioral sensitization and conditioned place preference. In the first test, an increase in locomotor activity is observed with intermittent drug exposure. The latter tests for the preference of a particular environment associated with drug exposure by measuring the time an animal spends in the compartment where a drug was received compared with the compartment where only saline was injected (conditioned place preference). Both tests have in common that they are sensitive to cue-conditioned effects of addictive drugs. Subsequent exposures to the environment without the drug lead to extinction of the place preference, which can be reinstated with a low dose of the drug or the presentation of a conditioned stimulus. These persistent changes serve as a model of relapse and have been linked to synaptic plasticity of excitatory transmission in the ventral tegmental area, nucleus accumbens, and prefrontal cortex (see also Box: The Dopamine Hypothesis of Addiction). More sophisticated tests rely on self-administration of the drug, in which a rat or a mouse has to press a lever in order to obtain an injection of, for example, cocaine. Once the animal has learned the association with a conditioned stimulus (eg, light or brief sound), the simple presentation of the cue elicits drug seeking. Prolonged self-administration of addictive drugs over months leads to behaviors in rats that closely resemble human addiction. Such “addicted” rodents are very strongly motivated to seek cocaine, continue looking for the drug even when no longer available, and self-administer cocaine in spite of negative consequences, such as an electric foot shock. These findings suggest that addiction is a disease that does not respect species boundaries.
DEPENDENCE: TOLERANCE & WITHDRAWAL
With chronic exposure to addictive drugs, the brain shows signs of adaptation. For example, if morphine is used at short intervals, the dose has to be progressively increased over the course of several days to maintain rewarding or analgesic effects. This phenomenon is called tolerance. It may become a serious problem because of increasing side effects—eg, respiratory depression—that do not show as much tolerance and may lead to fatalities associated with overdose.
Tolerance to opioids may be due to a reduction of the concentration of a drug or a shorter duration of action in a target system (pharmacokinetic tolerance). Alternatively, it may involve changes of μ-opioid receptor function (pharmacodynamic tolerance). In fact, many μ-opioid receptor agonists promote strong receptor phosphorylation that triggers the recruitment of the adaptor protein β-arrestin, causing G proteins to uncouple from the receptor and to internalize within minutes (see Chapter 2). Since this decreases signaling, it is tempting to explain tolerance by such a mechanism. However, morphine, which strongly induces tolerance, does not recruit β-arrestins and fails to promote receptor internalization. Conversely, other agonists that drive receptor internalization very efficiently induce only modest tolerance. Based on these observations, it has been hypothesized that desensitization and receptor internalization actually protect the cell from overstimulation. In this model, morphine, by failing to trigger receptor endocytosis, disproportionally stimulates adaptive processes, which eventually cause tolerance. Although the molecular identity of these processes is still under investigation, they may be similar to the ones involved in withdrawal (see below).
Adaptive changes become fully apparent once drug exposure is terminated. This state is called withdrawal and is observed to varying degrees after chronic exposure to most drugs of abuse. Withdrawal from opioids in humans is particularly strong (described below). Studies in rodents have added significantly to our understanding of the neural and molecular mechanisms that underlie dependence. For example, signs of dependence, as well as analgesia and reward, are abolished in knockout mice lacking the μ-opioid receptor, but not in mice lacking other opioid receptors (δ, κ). Although activation of the μ-opioid receptor initially strongly inhibits adenylyl cyclase, this inhibition becomes weaker after several days of repeated exposure. The reduction of the inhibition of adenylyl cyclase is due to a counter-adaptation of the enzyme system during exposure to the drug, which results in overproduction of cAMP during subsequent withdrawal. Several mechanisms exist for this adenylyl cyclase compensatory response, including up-regulation of transcription of the enzyme. Increased cAMP concentrations in turn strongly activate the transcription factor cyclic AMP response element binding protein (CREB), leading to the regulation of downstream genes. Of the few such genes identified to date, one of the most interesting is the gene for the endogenous κ-opioid ligand dynorphin. The main targets of dynorphin are the presynaptic κ-opioid receptors that regulate the release of dopamine in the nucleus accumbens.
ADDICTION: A DISEASE OF MALADAPTIVE MALADAPTIVE
Addiction is characterized by a high motivation to obtain and use a drug despite negative consequences. With time, drug use becomes compulsive (“wanting without liking”). Addiction is a recalcitrant, chronic, and stubbornly relapsing disease that is very difficult to treat.
The central problem is that even after successful withdrawal and prolonged drug-free periods, addicted individuals have a high risk of relapsing. Relapse is typically triggered by one of the following three conditions: re-exposure to the addictive drug, stress, or a context that recalls prior drug use. It appears that when paired with drug use, a neutral stimulus may undergo a switch and motivate (“trigger”) addiction-related behavior. This phenomenon may involve synaptic plasticity in the target nuclei of the mesolimbic projection (eg, projections from the medial prefrontal cortex to the neurons of the nucleus accumbens that express the D1 receptors). Several recent studies suggest that the recruitment of the dorsal striatum is responsible for the compulsion. This switch may depend on synaptic plasticity in the nucleus accumbens of the ventral striatum, where mesolimbic dopamine afferents converge with glutamatergic afferents to modulate their function. If dopamine release codes for the prediction error of reward (see Box: The Dopamine Hypothesis of Addiction), pharmacologic stimulation of the mesolimbic dopamine systems will generate an unusually strong learning signal. Unlike natural rewards, addictive drugs continue to increase dopamine even when reward is expected. Such overriding of the prediction error signal may eventually be responsible for the usurping of memory processes by addictive drugs.
The Dopamine Hypothesis of Addiction
In the earliest version of the hypothesis described in this chapter, mesolimbic dopamine was believed to be the neurochemical correlate of pleasure and reward. However, during the past decade, experimental evidence has led to several revisions. Phasic dopamine release may actually code for the prediction error of reward rather than the reward itself. This distinction is based on pioneering observations in monkeys that dopamine neurons in the ventral tegmental area (VTA) are most efficiently activated by a reward (eg, a few drops of fruit juice) that is not anticipated. When the animal learns to predict the occurrence of a reward (eg, by pairing it with a stimulus such as a sound), dopamine neurons stop responding to the reward itself (juice), but increase their firing rate when the conditioned stimulus (sound) occurs. Finally, if reward is predicted but not delivered (sound but no juice), dopamine neurons are inhibited below their baseline activity and become silent. In other words, the mesolimbic system continuously scans the reward situation. It increases its activity when reward is larger than expected, and shuts down in the opposite case, thus coding for the prediction error of reward.
Under physiologic conditions the mesolimbic dopamine signal could represent a learning signal responsible for reinforcing constructive behavioral adaptation (eg, learning to press a lever for food). Addictive drugs, by directly increasing dopamine, would generate a strong but inappropriate learning signal, thus hijacking the reward system and leading to pathologic reinforcement. As a consequence, behavior becomes compulsive; that is decisions are no longer planned and under control, but automatic, which is the hallmark of addiction.
This appealing hypothesis has been challenged based on the observation that some reward and drug-related learning is still possible in the absence of dopamine. Another intriguing observation is that mice genetically modified to lack the primary molecular target of cocaine, the dopamine transporter DAT, still self-administer the drug. Only when transporters of other biogenic amines are also knocked out does cocaine completely lose its rewarding properties. However, in DAT-/- mice, in which basal synaptic dopamine levels are high, cocaine still leads to increased dopamine release, presumably because other cocaine-sensitive monoamine transporters (NET, SERT) are able to clear some dopamine. When cocaine is given, these transporters are also inhibited and dopamine is again increased. As a consequence of this substitution among monoamine transporters, fluoxetine (a selective serotonin reuptake inhibitor, see Chapter 30) becomes addictive in DAT-/- mice. This concept is supported by newer evidence showing that deletion of the cocaine-binding site on DAT leaves basal dopamine levels unchanged but abolishes the rewarding effect of cocaine.
The dopamine hypothesis of addiction has also been challenged by the observation that salient stimuli that are not rewarding (they may actually even be aversive and therefore negative reinforcers) also activate a subpopulation of dopamine neurons in the VTA. The neurons that are activated by aversive stimuli preferentially project to the prefrontal cortex, while the dopamine neurons inhibited by aversive stimuli are those that mostly target the nucleus accumbens. These recent findings suggest that in parallel to the reward system, a system for aversion-learning originates in the VTA.
Regardless of the many roles of dopamine under physiologic conditions, all addictive drugs significantly increase its concentration in target structures of the mesolimbic projection. This suggests that high levels of dopamine may actually be at the origin of the adaptive changes that underlie dependence and addiction, a concept that is now supported by novel techniques that allow controlling the activity of dopamine neurons in vivo. In fact manipulations that drive sustained activity of VTA dopamine neurons cause the same cellular adaptations and behavioral changes typically observed with addictive drug exposure.
The involvement of learning and memory systems in addiction is also suggested by clinical studies. For example, the role of context in relapse is supported by the report that soldiers who became addicted to heroin during the Vietnam War had significantly better outcomes when treated after their return home, compared with addicts who remained in the environment where they had taken the drug. In other words, cravings may recur at the presentation of contextual cues (eg, people, places, or drug paraphernalia). Current research therefore focuses on the effects of drugs on associative forms of synaptic plasticity, such as long-term potentiation (LTP), which underlie learning and memory (see Box: Synaptic Plasticity & Addiction).
Long-term potentiation (LTP) is a form of experience-dependent synaptic plasticity that is induced by activating glutamate receptors of the N-methyl-D-aspartate (NMDA) type. Since NMDA receptors are blocked by magnesium at negative potentials, their activation requires the concomitant release of glutamate (presynaptic activity) onto a receiving neuron that is depolarized (postsynaptic activity). Correlated pre- and postsynaptic activity durably enhances synaptic efficacy and triggers the formation of new connections. Because associativity is a critical component, LTP has become a leading candidate mechanism underlying learning and memory. LTP can be elicited at glutamatergic synapses of the mesolimbic reward system and is modulated by dopamine. Drugs of abuse could therefore interfere with LTP at sites of convergence of dopamine and glutamate projections (eg, ventral tegmental area [VTA], nucleus accumbens, or prefrontal cortex). Interestingly, exposure to an addictive drug triggers a specific form of synaptic plasticity at excitatory afferents (drug-evoked synaptic plasticity) and potentiates GABAA receptor-mediated inhibition of the VTA GABA neurons. As a consequence, the excitability of dopamine neurons is increased, the synaptic calcium sources altered, and the rules for subsequent LTP inverted. In the nucleus accumbens, drug-evoked synaptic plasticity appears with some delay and mostly involves the D1 receptor-expressing neurons, which are the ones projecting back to the VTA to control the activity of the GABA neurons. Manipulations in mice that prevent or reverse drug-evoked plasticity in vivo also have effects on persistent changes of drug-associated behavioral sensitization or cue-induced drug seeking, providing more direct evidence for a causal role of synaptic plasticity in drug-adaptive behavior.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


