What causes degeneration of dopaminergic neurons? No one knows for sure. However, some evidence strongly implicates alpha-synuclein—a potentially toxic protein synthesized by dopaminergic neurons. Under normal conditions, alpha-synuclein is rapidly degraded. As a result, it doesn’t accumulate, and no harm occurs. Degradation of alpha-synuclein requires two other proteins: parkin and ubiquitin. (Parkin is an enzyme that catalyzes the binding of alpha-synuclein to ubiquitin. When bound to ubiquitin, alpha-synuclein can be degraded.) If any of these proteins—alpha-synuclein, parkin, or ubiquitin—is defective, degradation of alpha-synuclein cannot take place. When this occurs, alpha-synuclein accumulates inside the cell, forming neurotoxic fibrils. At autopsy, these fibrils are visible as Lewy bodies, which are characteristic of PD pathology. Failure to degrade alpha-synuclein appears to result from two causes: genetic vulnerability and toxins in the environment. Defective genes coding for all three proteins have been found in families with inherited forms of PD. In people with PD that is not inherited, environmental toxins may explain the inability to degrade alpha-synuclein.
As discussed in Chapter 24, movement disorders similar to those of PD can occur as side effects of antipsychotic drugs. These dyskinesias, which are referred to as extrapyramidal side effects, result from blockade of dopamine receptors in the striatum. This drug-induced parkinsonism can be managed with some of the drugs used to treat PD.
Overview of Motor Symptom Management
Therapeutic Goal
Ideally, treatment would reverse neuronal degeneration, or at least prevent further degeneration, and control symptoms. Unfortunately, the ideal treatment doesn’t exist: we have no drugs that can prevent neuronal damage or reverse damage that has already occurred. Drugs can only provide symptomatic relief; they do not cure PD. Furthermore, there is no convincing proof that any current drug can delay disease progression. Hence the goal of pharmacologic therapy is simply to improve the patient’s ability to carry out activities of daily living. Drug selection and dosage are determined by the extent to which PD interferes with work, walking, dressing, eating, bathing, and other activities. Drugs benefit the patient primarily by improving bradykinesia, gait disturbance, and postural instability. Tremor and rigidity, although disturbing, are less disabling.
Drugs Employed
Given the neurochemical basis of parkinsonism—too little striatal dopamine and too much acetylcholine—the approach to treatment is obvious: give drugs that can restore the functional balance between dopamine and acetylcholine. To accomplish this, two types of drugs are used: (1) dopaminergic agents (i.e., drugs that directly or indirectly activate dopamine receptors) and (2) anticholinergic agents (i.e., drugs that block receptors for acetylcholine). Of the two groups, dopaminergic agents are by far the more widely employed.
As shown in Table 17.1, dopaminergic drugs act by several mechanisms: levodopa is converted to dopamine, which activates dopamine receptors directly; inhibitors of monoamine oxidase-B (MAO-B) prevent dopamine breakdown; amantadine promotes dopamine release (and may also block dopamine reuptake); and the inhibitors of catechol-O-methyltransferase (COMT) enhance the effects of levodopa by blocking its degradation.
TABLE 17.1
Dopaminergic Agents for Parkinson Disease
| Drug | Mechanism of Action | Therapeutic Role |
| DOPAMINE REPLACEMENT | ||
| Levodopa/carbidopa | Levodopa undergoes conversion to DA in the brain and then activates DA receptors (carbidopa blocks destruction of levodopa in the periphery) | First-line drug, or supplement to a dopamine agonist |
| DOPAMINE AGONISTS | ||
| Nonergot Derivatives | Directly activate DA receptors | Pramipexole and ropinirole are first-line drugs, or supplements to levodopa. Apomorphine—a subQ nonergot agent—is reserved for rescue therapy during “off” times. Ergot derivatives are generally avoided. |
Apomorphine Pramipexole Ropinirole Rotigotine | ||
| Ergot Derivatives | ||
Bromocriptine Cabergoline | ||
| COMT INHIBITORS | ||
Entacapone Tolcapone | Inhibit breakdown of levodopa by COMT | Adjunct to levodopa to decrease “wearing off”; entacapone is more effective and safer than tolcapone |
| MAO-B INHIBITORS | ||
Rasagiline Selegiline | Inhibit breakdown of DA by MAO-B | Used in newly diagnosed patients and for managing “off” times during levodopa therapy |
| DOPAMINE RELEASER | ||
| Amantadine | Promotes release of DA from remaining DA neurons; may also block DA reuptake | May help reduce levodopa-induced dyskinesias |
In contrast to the dopaminergic drugs, which act by multiple mechanisms, all of the anticholinergic agents share the same mechanism: blockade of muscarinic receptors in the striatum.
Clinical Guidelines
Management of PD is generally under the guidance of a neurologist who oversees pharmacologic management. Other providers are responsible for differing aspects of care, including management of primary care needs and monitoring of patient status. Thus it is important for all providers involved in care to have a familiarity of drugs used to manage PD. The American Academy of Neurology (AAN) has developed a set of evidence-based guidelines for the treatment of PD and management of individual manifestations of the disease (see https://www.aan.com/Guidelines). The recommendations that follow are based on these guidelines. Lifespan considerations are summarized in the Patient-Centered Care Across the Lifespan box for Parkinson’s Disease.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN
Drugs for Parkinson Disease
| Life Stage | Considerations or Concerns |
| Children | Juvenile PD in patients younger than 18 years is extremely rare; therefore, many drugs for PD have not been tested in children. Only amantadine, benztropine, and bromocriptine have approval for pediatric populations. Selegiline is contraindicated in children younger than 12 years. |
| Pregnant women | Bromocriptine and cabergoline are FDA Pregnancy Risk Category B (although the manufacturer recommends stopping them after pregnancy is determined). All other drugs in this chapter are FDA Pregnancy Risk Category C owing to adverse events in animal studies. Of note, it is rare for a woman of childbearing age to develop PD. |
| Breastfeeding women | Bromocriptine and cabergoline interfere with lactation. Anticholinergics such as benztropine can suppress lactation. Breastfeeding is not recommended for women taking other drugs in this chapter. |
| Older adults | The average age of PD diagnosis is 62 years; therefore, most prescriptions are written for older adults. Adverse effects tend to be more common and more serious in these patients. Beers criteria designate anticholinergic drugs (e.g., benztropine and trihexyphenidyl) as potentially inappropriate for use in geriatric patients. |
Drug Selection
Initial Treatment
For patients with mild symptoms, treatment can begin with an MAO-B inhibitor. MAO-B inhibitors confer mild, symptomatic benefit.
For patients with more severe symptoms, treatment should begin with either levodopa (combined with carbidopa) or a dopamine agonist. Levodopa is more effective than the dopamine agonists, but long-term use carries a higher risk for disabling dyskinesias. Hence the choice must be tailored to the patient: if improving motor function is the primary objective, then levodopa is preferred. However, if drug-induced dyskinesias are a primary concern, then a dopamine agonist would be preferred.
Management of Motor Fluctuations
Long-term treatment with levodopa or dopamine agonists is associated with two types of motor fluctuations: “off” times (loss of symptom relief) and drug-induced dyskinesias (involuntary movements). “Off” times can be reduced with three types of drugs: dopamine agonists, COMT inhibitors, and MAO-B inhibitors. Evidence of efficacy is strongest for entacapone (a COMT inhibitor) and rasagiline (an MAO-B inhibitor). The only drug recommended for dyskinesias is amantadine.
Neuroprotection
To date, there is no definitive proof that any drug can protect dopaminergic neurons from progressive degeneration. However, although no drug has yet been proved to provide neuroprotective effects for people with PD, studies suggest that some drugs are promising. For example, MAO-B inhibitors have provided neuroprotective effects in animal studies. Similarly, dopamine agonists have demonstrated neuroprotective effects in laboratory studies. For both drug categories, however, clinical studies in humans have been inconclusive. A growing body of research with levodopa supports a likely role for neuroprotection; however, because some studies demonstrate toxic effects in patients with PD, the risks may outweigh the benefits when given for this purpose.
Pharmacology of the Drugs Used for Motor Symptoms
Levodopa
Levodopa was introduced in the 1960s and has been a cornerstone of PD treatment ever since. Unfortunately, although the drug is highly effective, beneficial effects diminish over time.
Use in Parkinson Disease
Beneficial Effects
Levodopa is the most effective drug for PD. At the beginning of treatment, about 75% of patients experience a 50% reduction in symptom severity. Levodopa is so effective, in fact, that a diagnosis of PD should be questioned if the patient fails to respond.
Full therapeutic responses may take several months to develop. Consequently, although the effects of levodopa can be significant, patients should not expect immediate improvement. Rather, they should be informed that beneficial effects are likely to increase steadily over the first few months.
In contrast to the dramatic improvements seen during initial therapy, long-term therapy with levodopa has been disappointing. Although symptoms may be well controlled during the first 2 years of treatment, by the end of year 5, ability to function may deteriorate to pretreatment levels. This probably reflects disease progression and not development of tolerance to levodopa.
Acute Loss of Effect
Acute loss of effect occurs in two patterns: gradual loss and abrupt loss. Gradual loss—“wearing off”—develops near the end of the dosing interval and simply indicates that drug levels have declined to a subtherapeutic value. Wearing off can be minimized in three ways: (1) shortening the dosing interval, (2) giving a drug that prolongs levodopa’s plasma half-life (e.g., entacapone), and (3) giving a direct-acting dopamine agonist.
Abrupt loss of effect, often referred to as the “on-off” phenomenon, can occur at any time during the dosing interval—even while drug levels are high. “Off” times may last from minutes to hours. Over the course of treatment, “off” periods are likely to increase in both intensity and frequency. Drugs that can help reduce “off” times are listed in Table 17.2. As discussed later, avoiding high-protein meals may also help.
TABLE 17.2
Drugs for Motor Complications of Levodopa Therapy
| Drug | Drug Class |
| DRUGS FOR “OFF” TIMES | |
| Definitely Effective | |
| Entacapone | COMT inhibitor |
| Rasagiline | MAO-B inhibitor |
| Probably Effective | |
| Rotigotine | DA agonist |
| Pramipexole | DA agonist |
| Ropinirole | DA agonist |
| Tolcapone | COMT inhibitor |
| Possibly Effective | |
| Apomorphine | DA agonist |
| Cabergoline | DA agonist |
| Selegiline | MAO-B inhibitor |
| DRUG FOR LEVODOPA-INDUCED DYSKINESIAS | |
| Amantadine | DA-releasing agent |
Mechanism of Action
Levodopa reduces symptoms by increasing dopamine synthesis in the striatum (Fig. 17.2). Levodopa enters the brain through an active transport system that carries it across the blood-brain barrier. When in the brain, the drug undergoes uptake into the remaining dopaminergic nerve terminals that remain in the striatum. After uptake, levodopa, which has no direct effects of its own, is converted to dopamine, its active form. As dopamine, levodopa helps restore a proper balance between dopamine and acetylcholine.
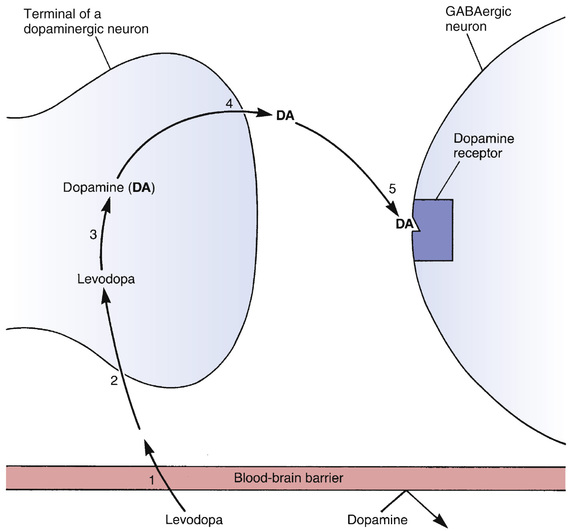
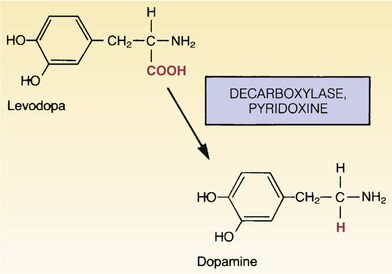
Conversion of levodopa to dopamine is depicted in Fig. 17.3. As indicated, the enzyme that catalyzes the reaction is called a decarboxylase (because it removes a carboxyl group from levodopa). The activity of decarboxylases is enhanced by pyridoxine (vitamin B6).
Why is PD treated with levodopa and not with dopamine itself? There are two reasons. First, dopamine cannot cross the blood-brain barrier (see Fig. 17.2). As noted, levodopa crosses the barrier by means of an active transport system, a system that does not transport dopamine. Second, dopamine has such a short half-life in the blood that it would be impractical to use even if it could cross the blood-brain barrier.
Pharmacokinetics
Levodopa is administered orally and undergoes rapid absorption from the small intestine. Food delays absorption by slowing gastric emptying. Furthermore, because neutral amino acids compete with levodopa for intestinal absorption (and for transport across the blood-brain barrier as well), high-protein foods will reduce therapeutic effects.
Only a small fraction of each dose reaches the brain. Most is metabolized in the periphery, primarily by decarboxylase enzymes and to a lesser extent by COMT. Peripheral decarboxylases convert levodopa into dopamine, an active metabolite. In contrast, COMT converts levodopa into an inactive metabolite. Like the enzymes that decarboxylate levodopa within the brain, peripheral decarboxylases work faster in the presence of pyridoxine. Because of peripheral metabolism, less than 2% of each dose enters the brain if levodopa is given alone. Fortunately, levodopa is now available only in combination preparations with either carbidopa or carbidopa and entacapone. These additional agents decrease the amount of decarboxylation in the periphery so that more of the drug can enter the CNS. This is discussed in greater detail later in the chapter.
Adverse Effects
Most side effects of levodopa are dose dependent. Older-adult patients, who are the primary users of levodopa, are especially sensitive to adverse effects.
Nausea and Vomiting
Most patients experience nausea and vomiting early in treatment. The cause is activation of dopamine receptors in the chemoreceptor trigger zone (CTZ) of the medulla. Nausea and vomiting can be reduced by administering levodopa in low initial doses and with meals. (Food delays levodopa absorption, causing a decrease in peak plasma drug levels and a corresponding decrease in stimulation of the CTZ.) However, because administration with food can reduce therapeutic effects by decreasing levodopa absorption, administration with meals should be avoided if possible. Giving additional carbidopa (without levodopa) can help reduce nausea and vomiting. Why carbidopa helps is unknown.
Dyskinesias
Ironically, levodopa, which is given to alleviate movement disorders, actually causes movement disorders in many patients. About 80% develop involuntary movements within the first year. Some dyskinesias are just annoying (e.g., head bobbing, tics, grimacing), whereas others can be disabling (e.g., ballismus, a rapid involuntary jerking or flinging of proximal muscle groups, or choreoathetosis, a slow involuntary writhing movement). These dyskinesias develop just before or soon after optimal levodopa dosage has been achieved. Dyskinesias can be managed in three ways. First, the dosage of levodopa can be reduced. However, dosage reduction may allow PD symptoms to reemerge. Second, we can give amantadine (see later), which can reduce dyskinesias in some patients. If these measures fail, the remaining options are usually surgery and electrical stimulation.
Cardiovascular Effects
Postural hypotension is common early in treatment. The underlying mechanism is unknown. Hypotension can be reduced by increasing intake of salt and water. An alpha-adrenergic agonist can help, too.
Conversion of levodopa to dopamine in the periphery can produce excessive activation of beta1 receptors in the heart. Dysrhythmias can result, especially in patients with heart disease.
Psychosis
Psychosis develops in about 20% of patients. Prominent symptoms are visual hallucinations, vivid dreams or nightmares, and paranoid ideation (fears of personal endangerment, sense of persecution, feelings of being followed or spied on). Activation of dopamine receptors is in some way involved. Symptoms can be reduced by lowering levodopa dosage, but this will reduce beneficial effects too.
Treatment of levodopa-induced psychosis with first-generation antipsychotics is problematic. Yes, these agents can decrease psychological symptoms. However, they will also intensify symptoms of PD because they block receptors for dopamine in the striatum. In fact, when first-generation antipsychotic agents are used for schizophrenia, the biggest problem is parkinsonian side effects, referred to as extrapyramidal symptoms (EPS).
Two second-generation antipsychotics—clozapine and quetiapine—have been used successfully to manage levodopa-induced psychosis. Unlike the first-generation antipsychotic drugs, clozapine and quetiapine cause little or no blockade of dopamine receptors in the striatum and hence do not cause EPS. In patients taking levodopa, these drugs can reduce psychotic symptoms without intensifying symptoms of PD. Clozapine and quetiapine are discussed in Chapter 24.
Central Nervous System Effects
Levodopa may cause a number of central nervous system (CNS) effects. These range from anxiety and agitation to memory and cognitive impairment. Insomnia and nightmares are common. Some patients experience problems with impulse control, resulting in behavioral changes associated with promiscuity, gambling, binge eating, or alcohol abuse.
Other Adverse Effects
Levodopa may darken sweat and urine; patients should be informed about this harmless effect. Some studies suggest that levodopa can activate malignant melanoma; however, others have failed to support this finding. Until more is known, it is important to perform a careful skin assessment of patients who are prescribed levodopa.
Drug Interactions
Interactions between levodopa and other drugs can (1) increase beneficial effects of levodopa, (2) decrease beneficial effects of levodopa, and (3) increase toxicity from levodopa. Major interactions are shown in Table 17.3. Several important interactions are discussed later.
TABLE 17.3
Major Drug Interactions of Levodopa
| Drug Category | Drug | Mechanism of Interaction |
| Drugs that increase beneficial effects of levodopa | Carbidopa | Inhibits peripheral decarboxylation of levodopa |
| Entacapone, tolcapone | Inhibit destruction of levodopa by COMT in the intestine and peripheral tissues | |
| Rotigotine, apomorphine, bromocriptine, cabergoline, pramipexole, ropinirole | Stimulate dopamine receptors directly and thereby add to the effects of dopamine derived from levodopa | |
| Amantadine | Promotes release of dopamine | |
| Anticholinergic drugs | Block cholinergic receptors in the CNS and thereby help restore the balance between dopamine and ACh | |
| Drugs that decrease beneficial effects of levodopa | Antipsychotic drugs* | Block dopamine receptors in the striatum |
| Drugs that increase levodopa toxicity | MAO inhibitors (especially nonselective MAO inhibitors) | Inhibition of MAO increases the risk for severe levodopa-induced hypertension |
First-Generation Antipsychotic Drugs
All of the first-generation antipsychotic drugs (e.g., chlorpromazine, haloperidol) block receptors for dopamine in the striatum. As a result, they can decrease therapeutic effects of levodopa. Accordingly, concurrent use of levodopa and these drugs should be avoided. As discussed previously, two second-generation agents—clozapine and quetiapine—do not block dopamine receptors in the striatum, so they can be used safely in patients with PD.
Monoamine Oxidase Inhibitors
Levodopa can cause a hypertensive crisis if administered to an individual taking a nonselective inhibitor of monoamine oxidase (MAO). The mechanism is as follows: (1) levodopa elevates neuronal stores of dopamine and norepinephrine (NE) by promoting synthesis of both transmitters. (2) Because intraneuronal MAO serves to inactivate dopamine and NE, inhibition of MAO allows elevated neuronal stores of these transmitters to grow even larger. (3) Because both dopamine and NE promote vasoconstriction, release of these agents in supranormal amounts can lead to massive vasoconstriction, thereby causing blood pressure to rise dangerously high. To avoid hypertensive crisis, nonselective MAO inhibitors should be withdrawn at least 2 weeks before giving levodopa.
Anticholinergic Drugs
As discussed previously, excessive stimulation of cholinergic receptors contributes to the dyskinesias of PD. Therefore, by blocking these receptors, anticholinergic agents can enhance responses to levodopa.
Pyridoxine
You may read advice to limit pyridoxine (vitamin B6). It is true that pyridoxine can decrease the amount of levodopa available to reach the CNS by accelerating decarboxylation of levodopa in the periphery. However, because levodopa is now always combined with carbidopa, a drug that suppresses decarboxylase activity, this potential interaction is no longer a clinical concern.
Food Interactions
High-protein meals can reduce therapeutic responses to levodopa. Neutral amino acids compete with levodopa for absorption from the intestine and for transport across the blood-brain barrier. Therefore a high-protein meal can significantly reduce both the amount of levodopa absorbed and the amount transported into the brain. It has been suggested that a high-protein meal could trigger an abrupt loss of effect (i.e., an “off” episode). Accordingly, patients should be advised to spread their protein consumption evenly throughout the day.
Preparations
At one time levodopa was available as a single drug. However, these single-drug preparations have been withdrawn from the market. Levodopa is now available only in combination preparations, either levodopa/carbidopa or levodopa/carbidopa/entacapone.
Levodopa/Carbidopa
The combination of levodopa plus carbidopa is our most effective therapy for PD. Levodopa plus carbidopa is available under three trade names: Rytary, Sinemet, and Duopa.
Mechanism of Action
Carbidopa has no therapeutic effects of its own; however, carbidopa inhibits decarboxylation of levodopa in the intestine and peripheral tissues, making more levodopa available to the CNS. Carbidopa does not prevent the conversion of levodopa to dopamine by decarboxylases in the brain because carbidopa is unable to cross the blood-brain barrier.
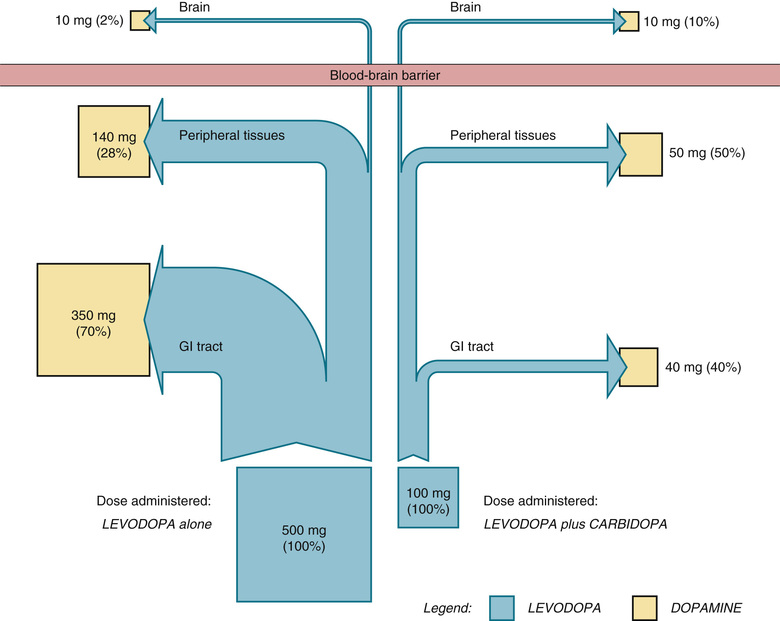
The effect of carbidopa is shown schematically in Fig. 17.4, which compares the fate of levodopa in the presence and absence of carbidopa. As mentioned previously, in the absence of carbidopa, about 98% of levodopa is lost in the periphery, leaving only 2% available to the brain. Why is levodopa lost? Primarily because decarboxylases in the gastrointestinal (GI) tract and peripheral tissues convert it to dopamine, which cannot cross the blood-brain barrier. When these decarboxylases are inhibited by carbidopa, only 90% of levodopa is lost in the periphery, leaving 10% for actions in the brain.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


