Diuretics Hydrochlorothiazide Spironolactone Beta-Adrenergic Blocker Metoprolol Inhibitors of the Renin-Angiotensin-Aldosterone System Captopril (angiotensin-converting enzyme inhibitor) Losartan (angiotensin II receptor blocker) Aliskiren (direct renin inhibitor) Eplerenone (aldosterone antagonist) Calcium Channel Blockers Verapamil Nifedipine
Review of Blood Pressure Control
Before discussing the antihypertensive drugs, we need to review the major mechanisms by which BP is controlled. This information will help you understand the mechanisms by which drugs lower BP.
Principal Determinants of Blood Pressure
The principal determinants of BP are shown in Fig. 39.1. As indicated, arterial pressure is the product of cardiac output and peripheral resistance. An increase in either will increase BP.
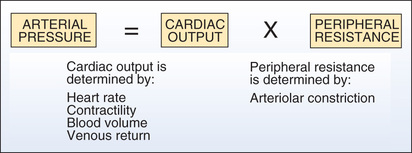
Cardiac Output.
Cardiac output is influenced by four factors: (1) heart rate, (2) myocardial contractility (force of contraction), (3) blood volume, and (4) venous return of blood to the heart. An increase in any of these will increase cardiac output, thereby causing BP to rise. Conversely, a decrease in these factors will make BP fall. Hence, to reduce BP, we might give a beta blocker to reduce cardiac output, or a diuretic to reduce blood volume, or a venodilator to reduce venous return.
Peripheral Vascular Resistance.
Vascular resistance is increased by arteriolar constriction. Accordingly, we can reduce BP with drugs that promote arteriolar dilation.
Systems that Help Regulate Blood Pressure
Having established that BP is determined by heart rate, myocardial contractility, blood volume, venous return, and arteriolar constriction, we can now examine how these factors are regulated. Three regulatory systems are of particular significance: (1) the sympathetic nervous system, (2) the renin-angiotensin-aldosterone system (RAAS), and (3) the kidney.
Sympathetic Baroreceptor Reflex.
The sympathetic nervous system employs a reflex circuit—the baroreceptor reflex—to keep BP at a preset level. This circuit operates as follows:
1. Baroreceptors in the aortic arch and carotid sinus sense BP and relay this information to the brainstem.
2. When BP is perceived as too low, the brainstem sends impulses along sympathetic nerves to stimulate the heart and blood vessels.
3. BP is then elevated by (a) activation of beta1 receptors in the heart, resulting in increased cardiac output; and (b) activation of vascular alpha1 receptors, resulting in vasoconstriction.
4. When BP has been restored to an acceptable level, sympathetic stimulation of the heart and vascular smooth muscle subsides.
The baroreceptor reflex frequently opposes our attempts to reduce BP with drugs. Opposition occurs because the “set point” of the baroreceptors is high in people with hypertension. That is, the baroreceptors are set to perceive excessively high BP as “normal” (i.e., appropriate). As a result, the system operates to maintain BP at pathologic levels. Consequently, when we attempt to lower BP using drugs, the reduced (healthier) pressure is interpreted by the baroreceptors as below what it should be, and, in response, signals are sent along sympathetic nerves to “correct” the reduction. These signals produce reflex tachycardia and vasoconstriction—responses that can counteract the hypotensive effects of drugs. Clearly, if treatment is to succeed, the regimen must compensate for the resistance offered by this reflex. Taking a beta blocker, which will block reflex tachycardia, can be an effective method of compensation. Fortunately, when BP has been suppressed with drugs for an extended time, the baroreceptors become reset at a lower level. Consequently, as therapy proceeds, sympathetic reflexes offer progressively less resistance to the hypotensive effects of medication.
Renin-Angiotensin-Aldosterone System.
The RAAS can elevate BP, negating the hypotensive effects of drugs. The RAAS is discussed in Chapter 36 and reviewed briefly here.
The RAAS elevates BP beginning with the release of renin from juxtaglomerular cells of the kidney. These cells release renin in response to reduced renal blood flow, reduced blood volume, reduced BP, and activation of beta1-adrenergic receptors on the cell surface. After its release, renin catalyzes the conversion of angiotensinogen into angiotensin I, a weak vasoconstrictor. After this, angiotensin-converting enzyme (ACE) acts on angiotensin I to form angiotensin II, a compound that constricts systemic and renal blood vessels. Constriction of systemic blood vessels elevates BP by increasing peripheral resistance. Constriction of renal blood vessels elevates BP by reducing glomerular filtration, which causes retention of salt and water, which in turn increases blood volume and BP. In addition to causing vasoconstriction, angiotensin II causes release of aldosterone from the adrenal cortex. Aldosterone acts on the kidney to further increase retention of sodium and water.
Because drug-induced reductions in BP can activate the RAAS, this system can counteract the effect we are trying to achieve. We have five ways to cope with this problem. First, we can suppress renin release with beta blockers. Second, we can prevent conversion of angiotensinogen to angiotensin I with a direct renin inhibitor (DRI). Third, we can prevent the conversion of angiotensin I into angiotensin II with an ACE inhibitor. Fourth, we can block receptors for angiotensin II with an angiotensin II receptor blocker (ARB). And fifth, we can block receptors for aldosterone with an aldosterone antagonist.
Renal Regulation of Blood Pressure.
As discussed in Chapter 34, the kidney plays a central role in long-term regulation of BP. When BP falls, glomerular filtration rate (GFR) falls too, thereby promoting retention of sodium, chloride, and water. The resultant increase in blood volume increases venous return to the heart, causing an increase in cardiac output, which in turn increases arterial pressure. We can neutralize renal effects on BP with diuretics.
Antihypertensive Mechanisms: Sites of Drug Action and Effects Produced
As discussed previously, drugs can lower BP by reducing heart rate, myocardial contractility, blood volume, venous return, and the tone of arteriolar smooth muscle. In this section we survey the principal mechanisms by which drugs produce these effects.
The major mechanisms for lowering BP are shown in Fig. 39.2 and Table 39.2. The figure depicts the principal sites at which antihypertensive drugs act. The table shows the effects elicited when drugs act at these sites. The numbering system used in the following text corresponds with the system used in Fig. 39.2 and Table 39.2.
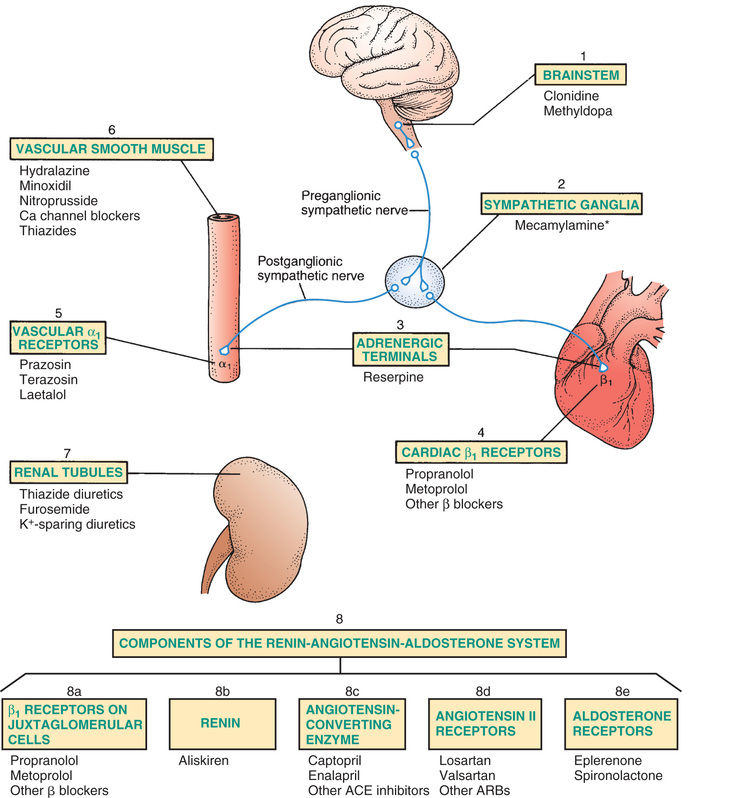
TABLE 39.2
Antihypertensive Effects Elicited by Drug Actions at Specific Sites
| Site of Drug Action* | Representative Drug | Drug Effects |
| Brainstem | Clonidine | Suppression of sympathetic outflow decreases sympathetic stimulation of the heart and blood vessels. |
| Sympathetic ganglia | Mecamylamine† | Ganglionic blockade reduces sympathetic stimulation of the heart and blood vessels. |
| Adrenergic nerve terminals | Reserpine | Reduced norepinephrine release decreases sympathetic stimulation of the heart and blood vessels. |
| Cardiac beta1 receptors | Metoprolol | Beta1 blockade decreases heart rate and myocardial contractility. |
| Vascular alpha1 receptors | Prazosin | Alpha1 blockade causes vasodilation. |
| Vascular smooth muscle | Hydralazine | Relaxation of vascular smooth muscle causes vasodilation. |
| Renal tubules | Hydrochlorothiazide | Promotion of diuresis decreases blood volume. |
| COMPONENTS OF THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM (8A TO 8E) | ||
| 8a.Beta1 receptors on juxtaglomerular cells | Metoprolol | Beta1 blockade suppresses renin release, resulting in (1) vasodilation secondary to reduced production of angiotensin II and (2) prevention of aldosterone-mediated volume expansion. |
| 8b.Renin | Aliskiren | Inhibition of renin suppresses formation of angiotensin I, which in turn decreases formation of angiotensin II and thereby reduces (1) vasoconstriction and (2) aldosterone-mediated volume expansion. |
| 8c.Angiotensin-converting enzyme (ACE) | Captopril | Inhibition of ACE decreases formation of angiotensin II and thereby prevents (1) vasoconstriction and (2) aldosterone-mediated volume expansion. |
| 8d.Angiotensin II receptors | Losartan | Blockade of angiotensin II receptors prevents angiotensin-mediated vasoconstriction and aldosterone-mediated volume expansion. |
| 8e.Aldosterone receptors | Eplerenone | Blockade of aldosterone receptors in the kidney promotes excretion of sodium and water and thereby reduces blood volume. |
1—Brainstem
Antihypertensive drugs acting in the brainstem suppress sympathetic outflow to the heart and blood vessels, resulting in decreased heart rate, decreased myocardial contractility, and vasodilation. Vasodilation contributes the most to reducing BP. Dilation of arterioles reduces BP by decreasing vascular resistance. Dilation of veins reduces BP by decreasing venous return to the heart.
2—Sympathetic Ganglia
Ganglionic blockade reduces sympathetic stimulation of the heart and blood vessels. Antihypertensive effects result primarily from dilation of arterioles and veins. Ganglionic blocking agents produce such a profound reduction in BP that they are used rarely, and then only for hypertensive emergencies. Because use is so limited, the last one available—mecamylamine—was voluntarily withdrawn from the U.S. market in 2009.
3—Terminals of Adrenergic Nerves
Antihypertensive agents that act at adrenergic nerve terminals decrease the release of norepinephrine, resulting in decreased sympathetic stimulation of the heart and blood vessels. These drugs, known as adrenergic neuron blocking agents, are used only rarely. In the United States reserpine is the only drug in this class still on the market.
4—Beta1-Adrenergic Receptors on the Heart
Blockade of cardiac beta1 receptors prevents sympathetic stimulation of the heart. As a result, heart rate and myocardial contractility decline.
5—Alpha1-Adrenergic Receptors on Blood Vessels
Blockade of vascular alpha1 receptors promotes dilation of arterioles and veins. Arteriolar dilation reduces peripheral resistance. Venous dilation reduces venous return to the heart.
6—Vascular Smooth Muscle
Several antihypertensive drugs (see Fig. 39.2) act directly on vascular smooth muscle to cause relaxation. One of these agents—sodium nitroprusside—is used only for hypertensive emergencies. The rest are used for chronic hypertension.
7—Renal Tubules
Diuretics act on renal tubules to promote salt and water excretion. As a result, blood volume declines, causing BP to fall.
Components of the RAAS (8a to 8e)
8a—Beta1 Receptors on Juxtaglomerular Cells.
Blockade of beta1 receptors on juxtaglomerular cells suppresses release of renin. The resultant decrease in angiotensin II levels has three effects: peripheral vasodilation, renal vasodilation, and suppression of aldosterone-mediated volume expansion.
8b—Renin.
Inhibition of renin decreases conversion of angiotensinogen into angiotensin I and thereby suppresses the entire RAAS. The result is peripheral vasodilation, renal vasodilation, and suppression of aldosterone-mediated volume expansion.
8c—Angiotensin-Converting Enzyme.
Inhibitors of ACE suppress formation of angiotensin II. The result is peripheral vasodilation, renal vasodilation, and suppression of aldosterone-mediated volume expansion.
8d—Angiotensin II Receptors.
Blockade of angiotensin II receptors prevents the actions of angiotensin II. Hence blockade results in peripheral vasodilation, renal vasodilation, and suppression of aldosterone-mediated volume expansion.
8e—Aldosterone Receptors.
Blockade of aldosterone receptors in the kidney promotes excretion of sodium and water and thereby reduces blood volume.
Classes of Antihypertensive Drugs
In this section we consider the principal drugs employed to treat chronic hypertension. Drugs for hypertensive emergencies and hypertensive disorders of pregnancy are considered separately.
Diuretics
Diuretics are a mainstay of antihypertensive therapy. These drugs reduce BP when used alone, and they can enhance the effects of other hypotensive drugs. The basic pharmacology of the diuretics is discussed in Chapter 35.
Thiazide Diuretics.
Thiazide diuretics (e.g., hydrochlorothiazide, chlorthalidone) are first-line drugs for hypertension. They reduce BP by two mechanisms: reduction of blood volume and reduction of arterial resistance. Reduced blood volume is responsible for initial antihypertensive effects. Reduced vascular resistance develops over time and is responsible for long-term antihypertensive effects. The mechanism by which thiazides reduce vascular resistance has not been determined.
Of the thiazides available, hydrochlorothiazide is used most widely. In fact, hydrochlorothiazide is used more widely than any other antihypertensive drug. Nonetheless, other thiazides, especially chlorthalidone, may be more effective.
The principal adverse effect of thiazides is hypokalemia. This can be minimized by consuming potassium-rich foods and using potassium supplements or a potassium-sparing diuretic. Other side effects include dehydration, hyperglycemia, and hyperuricemia.
Thiazides are superior to calcium channel blockers (CCBs) and ACE inhibitors as monotherapy and therefore are preferred.
Loop Diuretics.
Loop diuretics (e.g., furosemide) produce much greater diuresis than the thiazides. For most individuals with chronic hypertension, the amount of fluid loss that loop diuretics can produce is greater than needed or desirable. Consequently, loop diuretics are not used routinely for hypertension. Rather, they are reserved for (1) patients who need greater diuresis than can be achieved with thiazides and (2) patients with a low GFR (because thiazides won’t work when GFR is low). Like the thiazides, the loop diuretics lower BP by reducing blood volume and promoting vasodilation.
Most adverse effects are like those of the thiazides: hypokalemia, dehydration, hyperglycemia, and hyperuricemia. In addition, loop diuretics can cause hearing loss.
Potassium-Sparing Diuretics.
The degree of diuresis induced by the potassium-sparing agents (e.g., spironolactone) is small. Consequently, these drugs have only modest hypotensive effects. However, because of their ability to conserve potassium, these drugs can play an important role in an antihypertensive regimen. Specifically, they can balance potassium loss caused by thiazides or loop diuretics. The most significant adverse effect of the potassium-sparing agents is hyperkalemia. Because of the risk for hyperkalemia, potassium-sparing diuretics must not be used in combination with one another or with potassium supplements. Also, they should not be used routinely with ACE inhibitors, ARBs, or aldosterone antagonists, all of which promote significant hyperkalemia.
Sympatholytics (Antiadrenergic Drugs)
Sympatholytic drugs suppress the influence of the sympathetic nervous system on the heart, blood vessels, and other structures. These drugs are used widely for hypertension.
There are five subcategories of sympatholytic drugs: (1) beta blockers, (2) alpha1 blockers, (3) alpha/beta blockers, (4) centrally acting alpha2 agonists, and (5) adrenergic neuron blockers.
Beta-Adrenergic Blockers.
Like the thiazides, beta blockers (e.g., metoprolol) are widely used antihypertensive drugs. However, despite their efficacy and frequent use, the exact mechanism by which they reduce BP is somewhat uncertain. Beta blockers are less effective in black people than in whites.
The beta blockers have at least four useful actions in hypertension. First, blockade of cardiac beta1 receptors decreases heart rate and contractility, thereby causing cardiac output to decline. Second, beta blockers can suppress reflex tachycardia caused by vasodilators. Third, blockade of beta1 receptors on juxtaglomerular cells of the kidney reduces release of renin and thereby reduces angiotensin II−mediated vasoconstriction and aldosterone-mediated volume expansion. Fourth, long-term use of beta blockers reduces peripheral vascular resistance—by a mechanism that is unknown. This action could readily account for most of their antihypertensive effects.
Three beta blockers have intrinsic sympathomimetic activity: pindolol, penbutolol, and acebutolol. That is, they can produce mild activation of beta receptors while blocking receptor activation by strong agonists (e.g., norepinephrine). As a result, heart rate at rest is slowed less than with other beta blockers. Accordingly, if a patient develops symptomatic bradycardia with another beta blocker, switching to one of these may help.
Beta blockers can produce several adverse effects. Blockade of cardiac beta1 receptors can produce bradycardia, decreased atrioventricular (AV) conduction, and reduced contractility. Consequently, beta blockers should not be used by patients with sick sinus syndrome or second- or third-degree AV block—and must be used with care in patients with heart failure. Blockade of beta2 receptors in the lung can promote bronchoconstriction. Accordingly, beta blockers should be used with caution in patients with asthma. If an asthmatic individual must use a beta blocker, a beta1-selective agent (e.g., metoprolol) should be employed. Beta blockers can mask signs of hypoglycemia and therefore must be used with caution in patients with diabetes. Potential side effects of beta blockers include depression, insomnia, bizarre dreams, and sexual dysfunction; however, a review of older clinical trials has shown that the risk is small or nonexistent.
The basic pharmacology of the beta blockers is discussed in Chapter 14.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


