*Drug possesses more than one mechanism of action
Suppression of Sodium Influx
Before discussing AED actions, we need to review sodium channel physiology. Neuronal action potentials are propagated by influx of sodium through sodium channels, which are gated pores in the cell membrane that control sodium entry. For sodium influx to occur, the channel must be in an activated state. Immediately after sodium entry, the channel goes into an inactivated state, during which further sodium entry is prevented. Under normal circumstances, the inactive channel very quickly returns to the activated state, thereby permitting more sodium entry and propagation of another action potential.
Several AEDs, including phenytoin, carbamazepine, and lamotrigine, reversibly bind to sodium channels while they are in the inactivated state and thereby prolong channel inactivation. By delaying return to the active state, these drugs decrease the ability of neurons to fire at high frequency. As a result, seizures that depend on high-frequency discharge are suppressed.
Suppression of Calcium Influx
In axon terminals, influx of calcium through voltage-gated calcium channels promotes transmitter release. Hence drugs that block these calcium channels can suppress transmission. Ethosuximide acts by this mechanism.
Promotion of Potassium Efflux
During an action potential, influx of sodium causes neurons to depolarize, and then efflux of potassium causes neurons to repolarize. One AED—ezogabine (retigabine)—acts on voltage-gated potassium channels to facilitate potassium efflux. This action is believed to underlie the drug’s ability to slow repetitive neuronal firing and thereby provide seizure control.
Antagonism of Glutamate
Glutamic acid (glutamate) is the primary excitatory transmitter in the CNS. The compound works through two receptors: (1) N-methyl-D-aspartate (NMDA) receptors and (2) alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors. Perampanel is an AMPA glutamate receptor antagonist. Two other drugs—felbamate and topiramate—block the actions of glutamate at NMDA receptors and thereby suppress neuronal excitation.
Potentiation of Gamma-Aminobutyric Acid
Several AEDs potentiate the actions of GABA, an inhibitory neurotransmitter that is widely distributed throughout the brain. By augmenting the inhibitory influence of GABA, these drugs decrease neuronal excitability and thereby suppress seizure activity. Drugs increase the influence of GABA by several mechanisms. Benzodiazepines and barbiturates enhance the effects of GABA by mechanisms that involve direct binding to GABA receptors. Gabapentin promotes GABA release. Tiagabine inhibits GABA reuptake, and vigabatrin inhibits the enzyme that degrades GABA and thereby increases GABA availability.
Basic Therapeutic Considerations
Therapeutic Goal and Treatment Options
The goal in treating epilepsy is to reduce seizures to an extent that enables the patient to live a normal or nearly normal life. Ideally, treatment should eliminate seizures entirely. However, this may not be possible without causing intolerable side effects. Therefore we must balance the desire for complete seizure control against the acceptability of side effects.
Epilepsy may be treated with drugs or with nondrug therapies. As noted, drugs can benefit 60% to 70% of patients. This means that, of the 2.9 million Americans with epilepsy, between 870,000 and 1,160,000 cannot be treated successfully with drugs. For these people, nondrug therapy may well help. Three options exist: neurosurgery, vagus nerve stimulation, and the ketogenic diet. Of the three, neurosurgery has the best success rate, but vagus nerve stimulation is used most widely.
Diagnosis and Drug Selection
Control of seizures requires proper drug selection. As indicated in Table 19.1, many AEDs are selective for specific seizure disorders. Phenytoin, for example, is useful for treating tonic-clonic and partial seizures but not absence seizures. Conversely, ethosuximide is active against absence seizures but not against tonic-clonic or partial seizures. Only one drug—valproic acid—appears effective against practically all forms of epilepsy. Because most AEDs are selective for certain seizure disorders, effective treatment requires a proper match between the drug and the seizure. To make this match, the seizure type must be accurately diagnosed.
Making a diagnosis requires physical, neurologic, and laboratory evaluations along with a thorough history. The history should determine the age at which seizures began, the frequency and duration of seizure events, precipitating factors, and times when seizures occur. Physical and neurologic evaluations may reveal signs of head injury or other disorders that could underlie seizure activity, although in many patients the physical and neurologic evaluations may be normal. An electroencephalogram is essential for diagnosis. Other diagnostic tests that may be employed include computed tomography, positron emission tomography, and magnetic resonance imaging.
Pharmacologic management with AEDs is highly individualized. Very often, patients must try several AEDs before a regimen that is both effective and well tolerated can be established. Initial treatment should be done with just one AED. If this drug fails, it should be discontinued and a different AED should be tried. If this second drug fails, two options are open: (1) treatment with a third AED alone or (2) treatment with a combination of AEDs.
Drug Evaluation
After an AED has been selected, a trial period is needed to determine its effectiveness. During this time there is no guarantee that seizures will be controlled. Until seizure control is certain, the patient should be warned not to participate in driving and other activities that could be hazardous should a seizure occur.
Maintenance of a seizure frequency chart is important. The chart should be kept by the patient or a family member and should contain a complete record of all seizure events. This record can be used to help guide the provider in optimizing pharmacologic management.
During the process of drug evaluation, adjustments in dosage are often needed. No drug should be considered ineffective until it has been tested in sufficiently high dosages and for a reasonable time. Knowledge of plasma drug levels can be a valuable tool for establishing dosage and evaluating the effectiveness of a specific drug.
Monitoring Plasma Drug Levels
Safe and effective levels have been firmly established for most AEDs (see Table 19.2). Monitoring these levels can help guide dosage adjustments.
TABLE 19.2
Clinical Pharmacology of the Oral Antiepileptic Drugs
| Drug | Preparations | Product Name | Daily Dosinga | Daily Maintenance Dosagea | Target Serum Level (mcg/mL) | Induces Hepatic Drug Metabolism | |
| Adults (mg) | Children (mg/kg) | ||||||
| TRADITIONAL AEDS | |||||||
| Carbamazepine | IR tablets: 200 mg Chewable tablets: 100 mg ER tablets: 100, 200, 400 mg ER capsules: 100, 200, 300 mg Oral suspension: 20 mg/mL | Tegretol | 3 times | 800–1200 | 10–35 | 4–12 | Yes |
| Epitol | 3 times | ||||||
Tegretol XR, Tegretol CR  | 2 times | ||||||
| Carbatrol | 2 times | ||||||
| Equetro | 2 times | ||||||
| Ethosuximide | Capsules: 250 mg Syrup: 250 mg/5 mL | Zarontin | 1 or 2 times | 750 | 20 | 40–100 | No |
| Phenobarbital | Tablets: 15 mg, 16.2 mg, 30 mg, 32.4 mg, 60 mg, 64.8 mg, 97.2 mg, 100 mg Elixir: 20 mg/5 mL Oral solution: 20 mg/5 mL Solution for injection: 65 mg/mL, 130 mg/mL | Generic only | 1 or 2 times | 50–120 | 3–8 | 15–45 | Yes |
| Phenytoin | Chewable tablets: 50 mg Capsules: 30, 100, 200, 300 mg Oral suspension: 125 mg/5 mL Solution for injection: 50 mg/mL | Dilantin-125 | 2 or 3 times | 300–600 | 4–8 | 10–20 | Yes |
| Dilantin Infatab | 2 or 3 times | ||||||
| Phenytek (ER capsules) | 1 time | ||||||
| Dilantin (ER capsules) | 1 time | ||||||
| Fosphenytoin | Solution for injection: 100 mg PE/2 mL, 500 mg | ||||||
| Primidone | Tablets: 50 mg, 250 mg | Mysoline | 3 or 4 times | 500–750 | 10–25 | 5–12b | Yes |
| Valproic acid | See Table 19–4 | Depakene | 3 or 4 times | 500–3000 | 15–60 | 50–100 | No |
Depakote, Epival  | 3 or 4 times | ||||||
| Depakote ER | 2 times | ||||||
| Stavzor | 2 or 3 times | ||||||
| NEWER AEDS | |||||||
| Ezogabine | Tablets: 50 mg, 200 mg, 300 mg, 400 mg | Potiga | 3 times | 600–1200 | ND | ND | No |
| Felbamate | Tablets: 400 mg, 600 mg Oral suspension: 600 mg/5 mL | Felbatol | 3 or 4 times | 1200–3600 | 15–45 | ND | Yes |
| Gabapentin | Tablets: 600 mg, 800 mg Capsule: 100 mg, 300 mg, 400 mg, 600 mg, 800 mg Oral solution: 250 mg/5 mL | Neurontin | 3 times | 1200–3600 | 25–50 | 12–20 | No |
| Lacosamide | Tablets: 50 mg, 100 mg, 150 mg, 200 mg Oral solution: 10 mg/mL IV solution: 200 mg/20 mL | Vimpat | 2 times | 200–400 | ND | ND | No |
| Lamotrigine | Tablets: 25 mg, 100 mg, 150 mg, 200 mg Chewable tablets: 5 mg, 25 mg ODT: 25 mg, 50 mg, 100 mg, 200 mg ER tablet: 25 mg, 50 mg, 100 mg, 200 mg, 250 mg, 300 mg | Lamictal, Lamictal ODT | 2 times | 400–600c,d | 5c,d | 3–14 | No |
| Lamictal XR | 1 time | ||||||
| Levetiracetam | IR tablet: 250 mg, 500 mg, 750 mg, 1000 mg ER tablets: 500 mg, 750 mg ODT: 250 mg, 500 mg, 750 mg, 1000 mg Oral solution: 100 mg/mL IV solution: 500 mg/5 mL, 500 mg/100 mL, 1 g/100 mL, 1.5 g/100 mL | Keppra | 2 times | 2000–3000 | 40–100 | 10–40 | No |
| Keppra XR | 1 time | ||||||
| Oxcarbazepine | IR tablets: 150 mg, 300 mg, 600 mg ER tablets: 150 mg, 300 mg, 600 mg Oral suspension: 300 mg/5 mL | Trileptal | 2 times | 900–2400 | 30–46 | 3–40 | Yese |
| Oxtellar XR | 1 time | 1200–2400 | 20–29 kg: 900 mg/day; 29.1–39 kg: 1200 mg/day; >39 kg: 1800 mg/day | ||||
| Pregabalin | Capsule: 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 225 mg, 300 mg Oral solution: 20 mg/mL | Lyrica | 2 or 3 times | 150–600 | ND | ND | No |
| Rufinamide | Tablets: 200 mg, 400 mg Oral suspension: 40 mg/mL | Banzel | 2 times | 3200 | 45 | ND | Yesf |
| Tiagabine | Tablets: 2 mg, 4 mg, 12 mg, 16 mg | Gabitril | 2–4 times | 16–32 | 0.4d | ND | No |
| Topiramate | Tablets: 25 mg, 50 mg, 100 mg, 200 mg Sprinkle capsule: 15 mg, 25 mg ER sprinkle capsule: 25 mg, 50 mg, 100 mg, 150 mg, 200 mg ER capsule: 25 mg, 50 mg, 100 mg, 200 mg | Topamax, Trokendi XR, Qudexy XR | 2 times | 100–400 | 3–9 | 5–25 | No |
| Vigabatrin | Tablets: 500 mg Solution: 500 mg | Sabril | 2 times | 3000–6000 | 50–150 | ND | Yes |
| Zonisamide | Capsules: 25 mg, 50 mg, 100 mg | Zonegran | 1 or 2 times | 200–400 | 4–12 | 10–40 | No |
| Eslicarbazepine | Tablets: 400 mg, 600 mg, 800 mg | Aptiom | 1 time | 800–1600 | N/A | ND | Yes |
| Perampanel | Tablets: 2 mg, 4 mg, 6 mg, 8 mg, 10 mg, 12 mg | Fycompa | 1 time | 8–12 | N/A | N/D | No |
Monitoring plasma drug levels is especially helpful when treating major convulsive disorders (e.g., tonic-clonic seizures). Because these seizures can be dangerous, and because delay of therapy may allow the condition to worsen, rapid control of seizures is desirable. However, because these seizures occur infrequently, a long time may be needed to establish control if clinical outcome is relied on as the only means of determining an effective dosage. By adjusting initial doses on the basis of plasma drug levels (rather than on the basis of seizure control), we can readily achieve drug levels that are likely to be effective, thereby increasing our chances of establishing control quickly.
Measurements of plasma drug levels are less important for determining effective dosages for absence seizures. Because absence seizures occur very frequently (up to several hundred a day), observation of the patient is the best means for establishing an effective dosage: if seizures stop, dosage is sufficient; if seizures continue, it is likely that more drug is needed.
In addition to serving as a guide for dosage adjustment, knowledge of plasma drug levels can serve as an aid to (1) monitoring patient adherence, (2) determining the cause of lost seizure control, and (3) identifying causes of toxicity, especially in patients taking more than one drug.
Promoting Patient Adherence
Epilepsy is a chronic condition that requires regular and continuous therapy. As a result, seizure control is highly dependent on patient adherence. In fact, it is estimated that nonadherence accounts for about 50% of all treatment failures. Accordingly, promoting adherence should be a priority for all members of the health care team. Measures that can help include the following:
Withdrawing Antiepileptic Drugs
Some forms of epilepsy undergo spontaneous remission, and hence discontinuing treatment may eventually be appropriate. Unfortunately, there are no firm guidelines to indicate the most appropriate time to withdraw AEDs. However, after the decision to discontinue treatment has been made, agreement does exist on how drug withdrawal should be accomplished. The most important rule is that AEDs be withdrawn slowly (over a period of 6 weeks to several months). Failure to gradually reduce dosage is a frequent cause of SE. If the patient is taking two drugs to control seizures, they should be withdrawn sequentially, not simultaneously.
Suicide Risk With Antiepileptic Drugs
In 2008, the U.S. Food and Drug Administration (FDA) warned that all AEDs can increase suicidal thoughts and behavior. However, data gathered since 2008 suggest that the risk may be lower than previously believed and may apply only to certain AEDs.
Since the FDA issued its warning, other large studies have been conducted to clarify the relationship between AEDs and suicidality. Unfortunately, these studies have yielded conflicting results. Nonetheless, they do suggest three things. First, only some AEDs—especially topiramate and lamotrigine—are likely to increase suicidality, not all AEDs as warned by the FDA. Second, the risk for suicidal behavior may be related more to the illness than the medication: by analyzing data on 5,130,795 patients, researchers in the United Kingdom found that AEDs produced a small increase in suicidal behavior in patients with depression, but did not increase suicidal behavior in patients with epilepsy or bipolar disorder. And third, even if AEDs do promote suicidality, AED-related suicide attempts and completed suicides are very rare.
Given the uncertainty regarding AEDs and suicidality, what should the clinician do? Because epilepsy itself carries a risk for suicide, and because patients with epilepsy often have depression or anxiety (which increases the risk for suicide), prudence dictates screening all patients for suicide risk, whether or not AEDs increase that risk. In addition, after treatment begins, all patients should be monitored for increased anxiety, agitation, mania, and hostility—signs that may indicate the emergence or worsening of depression—and an increased risk for suicidal thoughts or behavior. Patients, families, and caregivers should be alerted to these signs and advised to report them immediately. Finally, two AEDs—topiramate and lamotrigine—should be used with special caution, given their significant association with suicidality.
Classification of Antiepileptic Drugs
The AEDs can be grouped into two major categories: traditional AEDs and newer AEDs. The traditional group has seven major members. The group of newer AEDs has 13 members. As shown in Table 19.3, both groups have their advantages and disadvantages. For example, clinical experience with the older AEDs is more extensive than with the newer ones, and the older drugs cost less. Both facts make the older drugs attractive. However, the older AEDs also have drawbacks, including troublesome side effects and complex drug interactions. Of importance, drugs in both groups appear equally effective—although few direct comparisons have been made. The bottom line? Neither group is clearly superior. Hence, when selecting an AED, drugs in both groups should be considered.
TABLE 19.3
Comparison of Traditional and Newer Antiepileptic Drugs
| Area of Comparison | AED Group | |
| Traditional AEDs* | Newer AEDs† | |
| Efficacy | Well established | Equally good (probably), but less well established |
| Clinical experience | Extensive | Less extensive |
| Therapeutic niche | Well established | Evolving |
| Tolerability | Less well tolerated | Better tolerated (usually) |
| Pharmacokinetics | Often complex | Less complex |
| Drug interactions | Extensive, owing to induction of drug-metabolizing enzymes | Limited, owing to little or no induction of drug-metabolizing enzymes |
| Safety in pregnancy | Less safe | Safer |
| Cost | Less expensive | More expensive |
Traditional Antiepileptic Drugs
The traditional AEDs have been in use for decades. Because of this extensive clinical experience, the efficacy and therapeutic niche of the traditional AEDs are well established. As a result, these drugs are prescribed more widely than the newer AEDs.
Although familiarity makes the traditional AEDs appealing, these drugs do have drawbacks. In general, they are less well tolerated than the newer AEDs, and they pose a greater risk to the developing fetus. Furthermore, owing to effects on drug-metabolizing enzymes (either induction or inhibition), they have complex interactions with other drugs, including other AEDs.
In the discussion that follows, we focus on the major traditional AEDs. They are phenytoin, fosphenytoin, carbamazepine, valproic acid, ethosuximide, phenobarbital, and primidone.
Phenytoin
Phenytoin [Dilantin, Phenytek] is our most widely used AED, despite having tricky kinetics and troublesome side effects. The drug is active against partial seizures as well as primary generalized tonic-clonic seizures. Phenytoin is of historical importance in that it was the first drug to suppress seizures without depressing the entire CNS. Consequently, phenytoin heralded the development of selective medications that could treat epilepsy while leaving most CNS functions undiminished.
Mechanism of Action
At the concentrations achieved clinically, phenytoin causes selective inhibition of sodium channels. Specifically, the drug slows recovery of sodium channels from the inactive state back to the active state. As a result, entry of sodium into neurons is inhibited, and hence action potentials are suppressed. Blockade of sodium entry is limited to neurons that are hyperactive. As a result, the drug suppresses activity of seizure-generating neurons while leaving healthy neurons unaffected.
Pharmacokinetics
Phenytoin has unusual pharmacokinetics that must be accounted for in therapy. Absorption varies substantially among patients. In addition, because of saturation kinetics, small changes in dosage can produce disproportionately large changes in serum drug levels. As a result, a dosage that is both effective and safe is difficult to establish.
Absorption
Absorption varies between the different oral formulations of phenytoin. With the oral suspension and chewable tablets absorption is relatively fast, whereas with the extended-release capsules absorption is delayed and prolonged.
In the past, there was concern that absorption also varied between preparations of phenytoin made by different manufacturers. However, it is now clear that all FDA-approved equivalent products have equivalent bioavailability. As a result, switching from one brand of phenytoin to another produces no more variability than switching between different lots of phenytoin produced by the same manufacturer.
Metabolism
The capacity of the liver to metabolize phenytoin is very limited. Doses of phenytoin needed to produce therapeutic effects are only slightly smaller than the doses needed to saturate the hepatic enzymes that metabolize phenytoin. Consequently, if phenytoin is administered in doses only slightly greater than those needed for therapeutic effects, the liver’s capacity to metabolize the drug will be overwhelmed, causing plasma levels of phenytoin to rise dramatically. This unusual relationship between dosage and plasma levels is illustrated in Fig. 19.1A. As you can see, after plasma levels have reached the therapeutic range, small changes in dosage produce large changes in plasma levels. As a result, small increases in dosage can cause toxicity, and small decreases can cause therapeutic failure. This relationship makes it difficult to establish and maintain a dosage that is both safe and effective.
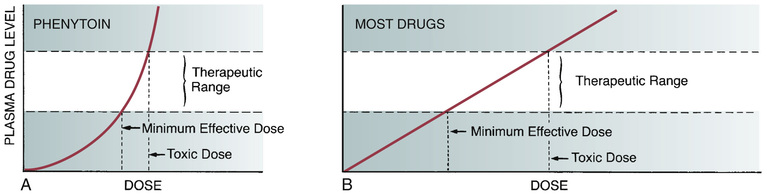
The relationship between dosage and plasma levels that exists for most drugs is detailed in Fig. 19.1B. As indicated, this relationship is linear, in contrast to the nonlinear relationship that exists for phenytoin. Accordingly, for most drugs, if the patient is taking doses that produce plasma levels that are within the therapeutic range, small deviations from that dosage produce only small deviations in plasma drug levels. Because of this relationship, with most drugs it is relatively easy to maintain plasma levels that are safe and effective.
Because of saturation kinetics, the half-life of phenytoin varies with dosage. At low doses, the half-life is relatively short—about 8 hours. However, at higher doses, the half-life becomes prolonged—in some cases up to 60 hours. At higher doses, there is more drug present than the liver can process. As a result, metabolism is delayed, causing the half-life to increase.
Therapeutic Uses
Epilepsy
Phenytoin can be used to treat all major forms of epilepsy except absence seizures. The drug is especially effective against tonic-clonic seizures, and phenytoin is a drug of choice for treating these seizures in adults and older children. (Carbamazepine is preferred to phenytoin for treating tonic-clonic seizures in young children.) Although phenytoin can be used to treat simple and complex partial seizures, the drug is less effective against these seizures than against tonic-clonic seizures. Phenytoin can be administered by intravenous (IV) injection to treat generalized convulsive SE, but other drugs are preferred.
Cardiac Dysrhythmias
Phenytoin is active against certain types of dysrhythmias. Antidysrhythmic applications are discussed in Chapter 41.
Adverse Effects
Effects on the CNS
Although phenytoin acts on the CNS in a relatively selective fashion to suppress seizures, the drug can still cause CNS side effects—especially when dosage is excessive. At therapeutic levels (10–20 mcg/mL), sedation and other CNS effects are mild. At plasma levels above 20 mcg/mL, toxicity can occur. Nystagmus (continuous back-and-forth movements of the eyes) is relatively common. Other manifestations of excessive dosage include sedation, ataxia (staggering gait), diplopia (double vision), and cognitive impairment.
Gingival Hyperplasia
Gingival hyperplasia (excessive growth of gum tissue) is characterized by swelling, tenderness, and bleeding of the gums. In extreme cases, patients require gingivectomy (surgical removal of excess gum tissue). Gingival hyperplasia is seen in about 20% of patients who take phenytoin. Can risk be reduced? Possibly. Risk may be minimized by good oral hygiene, including dental flossing and gum massage. Patients should be taught these techniques and encouraged to practice them. Supplemental folic acid is often recommended for this adverse effect based on studies that demonstrated that supplemental folic acid (0.5 mg/day) reduces gingival overgrowth. However, other research did not demonstrate this benefit. A subsequent Cochrane review established that only 15 of the studies met inclusion criteria and even those had issues with methodological quality. Their conclusion: Current evidence is insufficient to support the use of folic acid to prevent gingival hyperplasia so additional randomized control trial are needed.
Dermatologic Effects
Between 2% and 5% of patients develop a morbilliform (measles-like) rash. Rarely, morbilliform rash progresses to much more severe reactions: Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN). FDA labeling warns that the risk for developing SJS or TEN is strongly associated with a genetic mutation known as human leukocyte antigen (HLA)-B*1502, which occurs almost exclusively in people of Asian descent. For this reason, phenytoin should not be prescribed for patients known to have this mutation.
Effects in Pregnancy
Phenytoin is a teratogen. It can cause cleft palate, heart malformations, and fetal hydantoin syndrome, characterized by growth deficiency, motor or mental deficiency, microcephaly, craniofacial distortion, positional deformities of the limbs, hypoplasia of the nails and fingers, and impaired neurodevelopment. Because of these effects, phenytoin is classified in FDA Pregnancy Risk Category D and hence should be used during pregnancy only if safer alternatives are not effective and if the benefits of seizure control are deemed to outweigh the risk to the fetus.
Phenytoin can decrease synthesis of vitamin K–dependent clotting factors and can thereby cause bleeding tendencies in newborns. The risk for neonatal bleeding can be decreased by giving prophylactic vitamin K to the mother for 1 month before and during delivery and to the infant immediately after delivery.
Cardiovascular Effects
When phenytoin is administered by IV injection (to treat SE), cardiac dysrhythmias and hypotension may result. These dangerous responses can be minimized by injecting phenytoin no faster than 50 mg/min.
 Black Box Warning: Phenytoin [Dilantin, Phenytek] and Fosphenytoin [Cerebyx]
Black Box Warning: Phenytoin [Dilantin, Phenytek] and Fosphenytoin [Cerebyx]
When administered intravenously at rates exceeding 50 mg/min in adults, or at the slower rate of either 1 to 3 mg/kg/min or 50 mg/min in children, phenytoin can cause severe hypotension and cardiac dysrhythmias. Cardiac monitoring should be in place before administration.
Other Adverse Effects
Hirsutism (overgrowth of hair in unusual places) can be a disturbing response, especially in young women. Interference with vitamin D metabolism may cause rickets and osteomalacia (softening of the bones). Interference with vitamin K metabolism can lower prothrombin levels, thereby causing bleeding tendencies in newborns. Very rarely, liver damage may occur, probably because of drug allergy.
Drug Interactions
Phenytoin interacts with a large number of drugs. The more important interactions are discussed here.
Interactions Resulting From Induction of Hepatic Drug-Metabolizing Enzymes
Phenytoin stimulates synthesis of hepatic drug-metabolizing enzymes. As a result, phenytoin can decrease the effects of other drugs, including oral contraceptives, warfarin (an anticoagulant), and glucocorticoids (antiinflammatory and immunosuppressive drugs). Because avoiding pregnancy is desirable while taking antiseizure medications, and because phenytoin can decrease the effectiveness of oral contraceptives, the provider may need to increase the contraceptive dosage, or a switch to an alternative form of contraception may need to be made.
Drugs That Increase Plasma Levels of Phenytoin
Because the therapeutic range of phenytoin is narrow, slight increases in phenytoin levels can cause toxicity. Consequently, caution must be exercised when phenytoin is used with drugs that can increase its level. Drugs known to elevate phenytoin levels include diazepam (an antianxiety agent and AED), isoniazid (a drug for tuberculosis), cimetidine (a drug for gastric ulcers), and alcohol (when taken acutely). These agents increase phenytoin levels by reducing the rate at which phenytoin is metabolized. Valproic acid (an AED) elevates levels of free phenytoin by displacing phenytoin from binding sites on plasma proteins.
Drugs That Decrease Plasma Levels of Phenytoin
Carbamazepine, phenobarbital, and alcohol (when used chronically) can accelerate the metabolism of phenytoin, thereby decreasing its level. Breakthrough seizures can result.
CNS Depressants
The depressant effects of alcohol, barbiturates, and other CNS depressants will add with those of phenytoin. Advise patients to avoid alcohol and all other drugs with CNS-depressant actions.
Preparations, Dosage, and Administration
Preparations
There are a large number of phenytoin products on the market. Phenytoin products made by different manufacturers have equivalent bioavailability. Therefore, although switching between products from different manufacturers was a concern in the past, it is not a concern today.
Dosage
Dosing is highly individualized. Plasma drug levels are often monitored as an aid to establishing dosage. The dosing objective is to produce levels between 10 and 20 mcg/mL. Levels below 10 mcg/mL are too low to control seizures; levels above 20 mcg/mL produce toxicity. Because phenytoin has a relatively narrow therapeutic range (between 10 and 20 mcg/mL), and because of the nonlinear relationship between phenytoin dosage and phenytoin plasma levels, after a safe and effective dosage has been established, the patient should adhere to it rigidly.
When treatment is discontinued, dosage should be reduced gradually. Abrupt withdrawal may precipitate seizures.
Administration
Oral preparations may cause gastric discomfort. Patients should be informed that gastric upset can be reduced by administering phenytoin with or immediately after a meal. Patients using the oral suspension should shake it well before dispensing because failure to do so can result in uneven dosing.
Fosphenytoin
Fosphenytoin [Cerebyx] is a prodrug that is converted to phenytoin when metabolized. It is recommended as a substitute for oral phenytoin when the oral route is contraindicated. Because it is converted to phenytoin, the mechanism of action, therapeutic and adverse effects, and drug interactions are the same as those of phenytoin.
Pharmacokinetics
Pharmacokinetic properties are essentially the same as phenytoin after conversion. There are a few differences that are attributable to the prodrug in its nonhydrolyzed state.
Absorption
Fosphenytoin is available for parenteral administration only. Unlike phenytoin, fosphenytoin may be administered by the intramuscular (IM) route. This provides an advantage in that, if IV access is unattainable, the drug is rapidly absorbed after IM administration. Bioavailability is 100% using either the IM or the IV route.
Metabolism
Fosphenytoin is rapidly hydrolyzed to phenytoin. Its conversion half-life is 15 minutes.
Distribution
Fosphenytoin is 95% to 99% protein bound. Because it is more highly protein bound than phenytoin, protein binding with fosphenytoin may displace phenytoin from protein binding sites, resulting in a transient increase in free (unbound, active) phenytoin.
Adverse Effects
Adverse effects of fosphenytoin are the same as those of phenytoin, with one notable exception. During IV infusion, temporary paresthesias and itching, especially in the groin area, may occur. This infusion-related reaction will resolve when the infusion rate is decreased or within 10 minutes after completion of the infusion.
Dosage and Administration
Fosphenytoin has a unique dosing system. Although 150 mg of fosphenytoin will hydrolyze to 100 mg of phenytoin, rather than use standard milligram dosing, fosphenytoin is dosed in phenytoin equivalents (PE). Using this alternative, fosphenytoin 1 mg PE equals phenytoin 1 mg.
Unlike phenytoin, the IV formulation of fosphenytoin is compatible with standard IV solutions. When the drug is administered IM, the full dose may need to be divided into 2 to 4 separate injections.
Carbamazepine
Carbamazepine [Tegretol, Tegretol-XR, Tegretol CR  , Carbatrol, Epitol, Equetro] is a cornerstone of epilepsy therapy. The drug is active against partial seizures and tonic-clonic seizures but not absence seizures.
, Carbatrol, Epitol, Equetro] is a cornerstone of epilepsy therapy. The drug is active against partial seizures and tonic-clonic seizures but not absence seizures.
Mechanism of Action
Carbamazepine suppresses high-frequency neuronal discharge in and around seizure foci. The mechanism appears to be the same as that of phenytoin: delayed recovery of sodium channels from their inactivated state.
Pharmacokinetics
Absorption of carbamazepine is delayed and variable. Levels peak 4 to 12 hours after dosing. Overall bioavailability is about 80%. The drug distributes well to tissues.
Elimination is by hepatic metabolism. Carbamazepine is unusual in that its half-life decreases as therapy progresses. During the initial phase of treatment, the half-life is about 40 hours. With continued treatment, the half-life decreases to about 15 hours because carbamazepine, like phenytoin and phenobarbital, induces hepatic drug-metabolizing enzymes. By increasing its own metabolism, carbamazepine causes its own half-life to decline.
Therapeutic Uses
Epilepsy
Carbamazepine is effective against tonic-clonic, simple partial, and complex partial seizures. Because the drug causes fewer adverse effects than phenytoin and phenobarbital, it is often preferred to these agents. Many prescribers consider carbamazepine the drug of first choice for partial seizures. Carbamazepine is not effective against absence, myoclonic, or atonic seizures.
Bipolar Disorder
Carbamazepine can provide symptomatic control in patients with bipolar disorder (manic-depressive illness) and is often effective in patients who are refractory to lithium. The role of carbamazepine in bipolar disorder is discussed in Chapter 26.
Trigeminal and Glossopharyngeal Neuralgias
A neuralgia is a severe, stabbing pain that occurs along the course of a nerve. Carbamazepine can reduce neuralgia associated with the trigeminal and glossopharyngeal nerves. The mechanism is unknown. It should be noted that, although carbamazepine can reduce pain in these specific neuralgias, it is not generally effective as an analgesic and is not indicated for other kinds of pain.
Adverse Effects
CNS Effects
In contrast to phenytoin and phenobarbital, carbamazepine has minimal effects on cognitive function. This is a primary reason for selecting carbamazepine over other antiseizure drugs.
Carbamazepine can cause a variety of neurologic effects, including visual disturbances (nystagmus, blurred vision, diplopia), ataxia, vertigo, unsteadiness, and headache. These reactions are common during the first weeks of treatment, affecting 35% to 50% of patients. Fortunately, tolerance usually develops with continued use. These effects can be minimized by initiating therapy at low doses and giving the largest portion of the daily dose at bedtime.
Hematologic Effects
Carbamazepine-induced bone marrow suppression can cause leukopenia, anemia, and thrombocytopenia. However, serious reactions are rare. Thrombocytopenia and anemia, which have an incidence of 5%, respond to drug discontinuation. Leukopenia, which has an incidence of 10%, is usually transient and subsides even with continued drug use. Accordingly, carbamazepine should not be withdrawn unless the white blood cell count drops below 3000/mm3.
Fatal aplastic anemia has occurred during carbamazepine therapy. This reaction is extremely rare, having an incidence of 1 in 200,000.
To monitor for serious hematologic effects, complete blood counts should be performed before treatment and periodically thereafter. Patients with preexisting hematologic abnormalities should not use this drug. Patients should be informed about manifestations of hematologic abnormalities (fever, sore throat, pallor, weakness, infection, easy bruising, petechiae) and instructed to notify the prescriber if these occur.
Birth Defects
Carbamazepine is teratogenic. In humans, the drug is associated with a 2.6-fold increase in the risk for spina bifida, a neural tube defect. Because it can harm the fetus, carbamazepine is classified in FDA Pregnancy Risk Category D and hence should be used only if the benefits of seizure control are deemed to outweigh risks to the fetus.
Hypoosmolarity
Carbamazepine can inhibit renal excretion of water, apparently by promoting secretion of antidiuretic hormone. Water retention can reduce the osmolarity of blood and other body fluids, thereby posing a threat to patients with heart failure. Periodic monitoring of serum sodium levels is recommended.
Dermatologic Effects
Carbamazepine has been associated with several dermatologic effects, including morbilliform rash (10% incidence), photosensitivity reactions, SJS, and TEN. Mild reactions can often be treated with prednisone (an antiinflammatory agent) or an antihistamine. Severe reactions—SJS and TEN—necessitate drug withdrawal.
 Black Box Warning: Carbamazepine [Carbatrol, Epitol, Equetro, Tegretol]
Black Box Warning: Carbamazepine [Carbatrol, Epitol, Equetro, Tegretol]
Carbamazepine may cause serious skin reactions such as SJS and TEN. Fatalities may occur. The risk for a reaction is strongly associated with the HLA-B*1502 variant of the HLA-B gene that is found predominantly in people of Asian ancestry.
Aplastic anemia and agranulocytosis occurred in patients taking carbamazepine. Incidence is rare (±2–6 patients for each 1 million taking the drug.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


