Actions of Angiotensin II
Angiotensin II participates in all processes regulated by the RAAS. The most prominent actions of angiotensin II are vasoconstriction and stimulation of aldosterone release. Both actions raise blood pressure. In addition, angiotensin II (as well as aldosterone) can act on the heart and blood vessels to cause pathologic changes in their structure and function.
Vasoconstriction
Angiotensin II is a powerful vasoconstrictor. The compound acts directly on vascular smooth muscle (VSM) to cause contraction. Vasoconstriction is prominent in arterioles and less so in veins. As a result of angiotensin-induced vasoconstriction, blood pressure rises. In addition to its direct action on blood vessels, angiotensin II can cause vasoconstriction indirectly by acting on (1) sympathetic neurons to promote norepinephrine release, (2) the adrenal medulla to promote epinephrine release, and (3) the central nervous system to increase sympathetic outflow to blood vessels.
Release of Aldosterone
Angiotensin II acts on the adrenal cortex to promote synthesis and secretion of aldosterone, whose actions are discussed later. The adrenal cortex is highly sensitive to angiotensin II, and hence angiotensin II can stimulate aldosterone release even when angiotensin II levels are too low to induce vasoconstriction. Aldosterone secretion is enhanced when sodium levels are low and when potassium levels are high.
Alteration of Cardiac and Vascular Structure
Angiotensin II may cause pathologic structural changes in the heart and blood vessels. In the heart, it may cause hypertrophy and remodeling. In hypertension, angiotensin II may be responsible for increasing the thickness of blood vessel walls. In atherosclerosis, it may be responsible for thickening the intimal surface of blood vessels. And in heart failure and MI, it may be responsible for causing cardiac hypertrophy and fibrosis. Known effects of angiotensin II that could underlie these pathologic changes include the following:
Actions of Aldosterone
Regulation of Blood Volume and Blood Pressure
After being released from the adrenal cortex, aldosterone acts on distal tubules of the kidney to cause retention of sodium and excretion of potassium and hydrogen. Because retention of sodium causes water to be retained as well, aldosterone increases blood volume, which causes blood pressure to rise.
Pathologic Cardiovascular Effects
Until recently, knowledge of aldosterone’s actions was limited to effects on the kidney. Now, however, we know that aldosterone can cause more harmful effects. Like angiotensin II, aldosterone can promote cardiac remodeling and fibrosis. In addition, aldosterone can activate the sympathetic nervous system and suppress uptake of norepinephrine in the heart, thereby predisposing the heart to dysrhythmias. Also, aldosterone can promote vascular fibrosis (which decreases arterial compliance), and it can disrupt the baroreceptor reflex. These adverse effects appear to be limited to states such as heart failure, in which levels of aldosterone can be extremely high.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN
RAAS Inhibitors
| Life Stage | Patient Care Concerns |
| Infants | Captopril and enalapril have been used in infants safely for management of hypertension (HTN). |
| Children/adolescents | Some ACE inhibitors and ARBs are approved for use in children older than 6 years for treatment of HTN. |
| Pregnant women | Animal studies revealed that drugs that block the RAAS should be avoided in pregnancy, especially in the second and third trimesters. ACE inhibitors, ARBs, and DRIs are classified in FDA Pregnancy Risk Category D. |
| Breastfeeding women | Data are lacking regarding effects on the infant when breastfeeding. Caution is advised. |
| Older adults | The SCOPE and LIFE trials revealed a 25% decrease in stroke in patients 55 to 80 years old using losartan compared with atenolol. A 20% decreased risk for new-onset diabetes was seen with candesartan compared with placebo. |
Formation of Angiotensin II by Renin and Angiotensin-Converting Enzyme
Angiotensin II is formed through two sequential reactions. The first is catalyzed by renin, the second by ACE.
Renin
Renin catalyzes the formation of angiotensin I from angiotensinogen. This reaction is the rate-limiting step in angiotensin II formation. Renin is produced by juxtaglomerular cells of the kidney and undergoes controlled release into the bloodstream, where it cleaves angiotensinogen into angiotensin I.
Regulation of Renin Release
Because renin catalyzes the rate-limiting step in angiotensin II formation, and because renin must be released into the blood in order to act, the factors that regulate renin release regulate the rate of angiotensin II formation.
Release of renin can be triggered by multiple factors (see Fig. 36.1). Release increases in response to a decline in blood pressure, blood volume, plasma sodium content, or renal perfusion pressure. Reduced renal perfusion pressure is an especially important stimulus for renin release and can occur in response to (1) stenosis of the renal arteries, (2) reduced systemic blood pressure, and (3) reduced plasma volume (brought on by dehydration, hemorrhage, or chronic sodium depletion). For the most part, these factors increase renin release through effects exerted locally in the kidney. However, some of these factors may also promote renin release through activation of the sympathetic nervous system. (Sympathetic nerves increase secretion of renin by causing stimulation of beta1-adrenergic receptors on juxtaglomerular cells.)
Release of renin is suppressed by factors opposite to those that cause release. That is, renin secretion is inhibited by elevation of blood pressure, blood volume, and plasma sodium content. Hence, as blood pressure, blood volume, and plasma sodium content increase in response to renin release, further release of renin is suppressed. In this regard, we can view release of renin as being regulated by a classical negative feedback loop.
Angiotensin-Converting Enzyme (Kinase II)
ACE catalyzes the conversion of angiotensin I (inactive) into angiotensin II (highly active). ACE is located on the luminal surface of all blood vessels. The vasculature of the lungs is especially rich in the enzyme. Because ACE is abundant, conversion of angiotensin I into angiotensin II occurs almost instantaneously after angiotensin I has been formed. ACE is a relatively nonspecific enzyme that can act on a variety of substrates in addition to angiotensin I.
Nomenclature regarding ACE can be confusing and requires comment. As just noted, ACE can act on several substrates. When the substrate is angiotensin I, we refer to the enzyme as ACE. However, when the enzyme is acting on other substrates, we refer to it by different names. Of importance to us, when the substrate is a hormone known as bradykinin, we refer to the enzyme as kinase II. So, please remember, whether we call it ACE or kinase II, we’re talking about the same enzyme.
Regulation of Blood Pressure by the Renin-Angiotensin-Aldosterone System
The RAAS is poised to help regulate blood pressure. Factors that lower blood pressure turn the RAAS on; factors that raise blood pressure turn it off. However, although the RAAS does indeed contribute to blood pressure control, its role in normovolemic, sodium-replete individuals is only modest. In contrast, the system can be a major factor in maintaining blood pressure in the presence of hemorrhage, dehydration, or sodium depletion.
The RAAS, acting through angiotensin II, raises blood pressure through two basic processes: vasoconstriction and renal retention of water and sodium. Vasoconstriction raises blood pressure by increasing total peripheral resistance; retention of water and sodium raises blood pressure by increasing blood volume. Vasoconstriction occurs within minutes to hours of activating the system and hence can raise blood pressure quickly. In contrast, days, weeks, or even months are required for the kidney to raise blood pressure by increasing blood volume.
Angiotensin II acts in two ways to promote renal retention of water. First, by constricting renal blood vessels, angiotensin II reduces renal blood flow and thereby reduces glomerular filtration. Second, angiotensin II stimulates release of aldosterone from the adrenal cortex. Aldosterone then acts on renal tubules to promote retention of sodium and water and excretion of potassium.
Tissue (Local) Angiotensin II Production
In addition to the traditional RAAS that we’ve been discussing, in which angiotensin II is produced in the blood and then carried to target tissues, angiotensin II is produced in individual tissues. This permits discrete, local effects of angiotensin II independent of the main system. Interference with local production of angiotensin II may underlie some effects of the ACE inhibitors.
It is important to note that some angiotensin II is produced by pathways that do not involve ACE. As a result, drugs that inhibit ACE cannot completely block angiotensin II production.
Angiotensin-Converting Enzyme Inhibitors
The ACE inhibitors are important drugs for treating hypertension, heart failure, diabetic nephropathy, and MI. In addition, they are used to prevent adverse cardiovascular events in patients at risk. Their most prominent adverse effects are cough, angioedema, first-dose hypotension, and hyperkalemia. For all of these agents, beneficial effects result largely from suppressing formation of angiotensin II. Because the similarities among ACE inhibitors are much more striking than their differences, we will discuss these drugs as a group, rather than selecting a prototype to represent them.
Mechanism of Action and Overview of Pharmacologic Effects
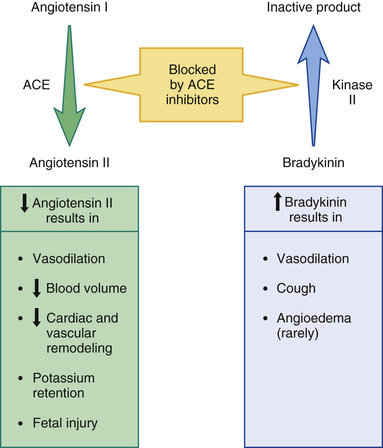
As shown in Fig. 36.2, ACE inhibitors produce their beneficial effects and adverse effects by (1) reducing levels of angiotensin II (through inhibition of ACE) and (2) increasing levels of bradykinin (through inhibition of kinase II). By reducing levels of angiotensin II, ACE inhibitors can dilate blood vessels (primarily arterioles and to a lesser extent veins), reduce blood volume (through effects on the kidney), and, importantly, prevent or reverse pathologic changes in the heart and blood vessels mediated by angiotensin II and aldosterone. Inhibition of ACE can also cause hyperkalemia and fetal injury. Elevation of bradykinin causes vasodilation (secondary to increased production of prostaglandins and nitric oxide) and can also promote cough and angioedema.
Pharmacokinetics
Regarding pharmacokinetics, the following generalizations apply:
• Nearly all ACE inhibitors are administered orally. The only exception is enalaprilat (the active form of enalapril), which is given intravenously.
• Except for captopril and moexipril, all oral ACE inhibitors can be administered with food.
• With the exception of captopril, all ACE inhibitors have prolonged half-lives and hence can be administered just once or twice a day. Captopril is administered 2 or 3 times a day.
• With the exception of lisinopril, all ACE inhibitors are prodrugs that must undergo conversion to their active form in the small intestine and liver. Lisinopril is active as given.
• All ACE inhibitors are excreted by the kidneys. As a result, nearly all can accumulate to dangerous levels in patients with kidney disease and hence dosages must be reduced in these patients. Only one agent—fosinopril—does not require a dosage reduction.
Therapeutic Uses
When the ACE inhibitors were introduced, their only indication was hypertension. Today, they are also used for heart failure, acute MI, left ventricular (LV) dysfunction, and diabetic and nondiabetic nephropathy. In addition, they can help prevent MI, stroke, and death in patients at high risk for cardiovascular events. It should be noted that no single ACE inhibitor is approved for all of these conditions (Table 36.1). However, given that all ACE inhibitors are very similar, it seems likely that all may produce similar benefits.
TABLE 36.1
ACE Inhibitors: Approved Indications and Adult Dosages
| Generic Name | Trade Name | Approved Indications | Availability | Starting Dosage* | Usual Maintenance Dosage* |
| Benazepril | Lotensin | Hypertension | 5-, 10-, 20-, 40-mg tablets | 10 mg once/day | 20–80 mg/day in 1 or 2 doses |
| Captopril | Capoten | Hypertension | 12.5-, 25-, 50-, 100-mg tablets | 25 mg 2 or 3 times/day | 25–50 mg 2 or 3 times/day |
| Heart failure | 6.25–12.5 mg 3 times/day | 50 mg 3 times/day | |||
| LVD after MI | 12.5 mg 3 times/day | 50 mg 3 times/day | |||
| Diabetic nephropathy | 25 mg 3 times/day | 25 mg 3 times/day | |||
| Enalapril | Vasotec | Hypertension | 2.5-, 5-, 10-, 20-mg tablets | 2.5–5 mg once/day | 10–40 mg/day in 1 or 2 doses |
| Heart failure | 2.5 mg twice/day | 10–20 mg twice/day | |||
| Asymptomatic LVD | 2.5 mg twice/day | 10 mg twice/day | |||
| Fosinopril | Monopril | Hypertension | 10-, 20-, 40-mg tablets | 10 mg once/day | 20–40 mg/day in 1 or 2 doses |
| Heart failure | 5–10 mg once/day | 20–40 mg once/day | |||
| Lisinopril | Prinivil, Zestril | Hypertension | 2.5-, 5-, 10-, 20-, 30-, 40-mg tablets | 10 mg once/day | 10–40 mg once/day |
| Heart failure | 2.5–5 mg once/day | 20–40 mg once/day | |||
| Acute MI | 5 mg once/day | 10 mg once/day | |||
| Moexipril | Univasc | Hypertension | 7.5-, 15-mg tablets | 7.5 mg once/day | 7.5–30 mg/day in 1 or 2 doses |
| Perindopril | Aceon, Coversyl  | Hypertension | 2-, 4-, 8-mg tablets | 4 mg once/day | 4–8 mg/day in 1 or 2 doses |
| Stable CAD | 4 mg once/day | 8 mg once/day | |||
| Quinapril | Accupril | Hypertension | 5-, 10-, 20-, 40-mg tablets | 10–20 mg/day | 20–80 mg/day in 1 or 2 doses |
| Heart failure | 5 mg twice/day | 20–40 mg twice/day | |||
| Ramipril | Altace | Hypertension | 1.25-, 2.5-, 5-, 10-mg capsules | 2.5 mg once/day | 2.5–20 mg/day in 1 or 2 doses |
| Heart failure after MI | 1.25–2.5 mg twice/day | 5 mg twice/day | |||
| Prevention of MI, stroke, and death in people at high risk for CVD | 2.5 mg/day for 1 wk | 10 mg once/day | |||
| Trandolapril | Mavik | Hypertension | 1-, 2-, 4-mg tablets | 1 mg once/day | 2–4 mg once/day |
| Heart failure after MI | 1 mg once/day | 4 mg once/day | |||
| LVD after MI | 1 mg once/day | 4 mg once/day |
Hypertension
All ACE inhibitors are approved for hypertension. These drugs are especially effective against malignant hypertension and hypertension secondary to renal arterial stenosis. They are also useful against essential hypertension of mild to moderate intensity—although maximal benefits may take several weeks to develop.
In patients with essential hypertension, the mechanism underlying blood pressure reduction is not fully understood. Initial responses are proportional to circulating angiotensin II levels and are clearly related to reduced formation of that compound. (By lowering angiotensin II levels, ACE inhibitors dilate blood vessels and reduce blood volume; both actions help lower blood pressure.) However, with prolonged therapy, blood pressure often undergoes additional decline. During this phase, there is no relationship between reductions in blood pressure and reductions in circulating angiotensin II. It may be that the delayed response is due to reductions in local angiotensin II levels—reductions that would not be revealed by measuring angiotensin II in the blood.
ACE inhibitors offer several advantages over most other antihypertensive drugs. In contrast to the sympatholytic agents, ACE inhibitors do not interfere with cardiovascular reflexes. Hence exercise capacity is not impaired, and orthostatic hypotension is minimal. In addition, these drugs can be used safely in patients with bronchial asthma, a condition that precludes the use of beta2-adrenergic antagonists. ACE inhibitors do not promote hypokalemia, hyperuricemia, or hyperglycemia—side effects seen with thiazide diuretics. Furthermore, they do not induce lethargy, weakness, or sexual dysfunction—responses that are common with other antihypertensive agents. Most important, ACE inhibitors reduce the risk for cardiovascular mortality caused by hypertension. The only other drugs proved to reduce hypertension-associated mortality are beta blockers and diuretics (see Chapter 39).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


