Drug Transporters
LEARNING OBJECTIVES
INTRODUCTION
Transporters are membrane proteins that are ubiquitous throughout the body functioning to control the influx of essential nutrients and ions and the efflux of cellular waste, toxins, and drugs. Transporters located in the liver, intestine, kidney, and blood–brain barrier (BBB) are of particular interest in drug development and utilization. Multiple transporters work in a coordinated fashion to transport endogenous and exogenous substances into and out of cells. In certain organs, such as the liver and intestine, transporters and drug-metabolizing enzymes work together to affect the pharmacokinetics of drugs.
Transporters play an important role in drug disposition, therapeutic efficacy, adverse drug reactions, and drug–drug interactions. Multiple environmental and genetic factors may affect the variability of transporter expression. Understanding the contribution of genetic variability to transporter expression and phenotype may allow clinicians to individualize drug therapy based on genetic factors. There are two major superfamilies of transporters important in drug disposition: solute carrier (SLC) transporters, a family of passive and active transporters that rely on chemical and/or electrical gradients for transport, and ATP-binding cassette (ABC) transporters, a family of primary active transporters that are ATP dependent. The following drug transporters will be discussed in this chapter with respect to the current knowledge of their pharmacogenetics: P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), organic anion-transporting polypeptide (OATP1B1), bile salt export pump (BSEP), organic cation transporter 1 (OCT1), organic cation transporter 2 (OCT2), multidrug and toxin extrusion 1 (MATE1), and organic anion transporters 1 and 3 (OAT1 and OAT3). For a review of other transporters not covered in this chapter, the reader is referred to other references.1,2 For most drug transporters, clinical evidence is sparse or, in the case of P-gp, conflicting. Due to the complex nature of drug disposition and subsequent effect on clinical outcome, isolating clinically significant drug transporter genetic variants is a challenging endeavor.
TRANSPORTERS IN THE INTESTINE
Most orally administered drugs are absorbed in the small intestine. Although the process of drug absorption is largely a passive process, transporters in the small intestine are determinants of the extent and variability of drug absorption that ultimately affect the pharmacokinetics of numerous drugs. Transporters expressed at the apical membrane of enterocytes may facilitate intestinal drug absorption or reduce the bioavailability of a drug. The resulting pharmacokinetic impact is dependent on the mechanism of transport. Transporters from both the ABC and SLC superfamilies are present in the apical membrane of the enterocyte (Figure 7–1). ABC transporters expressed on the apical membrane of the enterocyte act to limit drug absorption, where they transport drug from the enterocyte back into the intestinal lumen. The best characterized and apically localized efflux transporter is P-gp (ABCB1), but multidrug resistance protein 2 (MRP2/ABCC2) and BCRP (ABCG2) are also localized to the apical membrane and act to limit drug absorption. An important SLC transporter expressed on the apical membrane is PEPT1 (SLC15A1). PEPT1 is a nutrient transporter responsible for the absorption of dipeptides and tripeptides. Drugs that sufficiently resemble small peptides (peptidomimetics) may experience a faster rate of absorption than predicted because they are actively transported into the body in addition to being passively absorbed. Other SLC transporters are also localized to the apical membrane, but their role in facilitating the absorption of drugs is not established at this time. The impact of intestinal transporters on the absorption of drugs varies and depends on the passive permeability of the administered drug and the dose, among other factors. High passive permeability of drug will limit the role of transporters in drug absorption because the fraction of drug flux mediated by the transporter, either absorptive or efflux, will be small relative to the passive component. Drugs with low intrinsic passive permeability are more subject to manipulation by transporters. Similarly, a high dose of drug may saturate intestinal transport processes minimizing their impact. Because the passive component (diffusion) is not saturable, once transport is saturated absorption increases linearly with drug concentration.
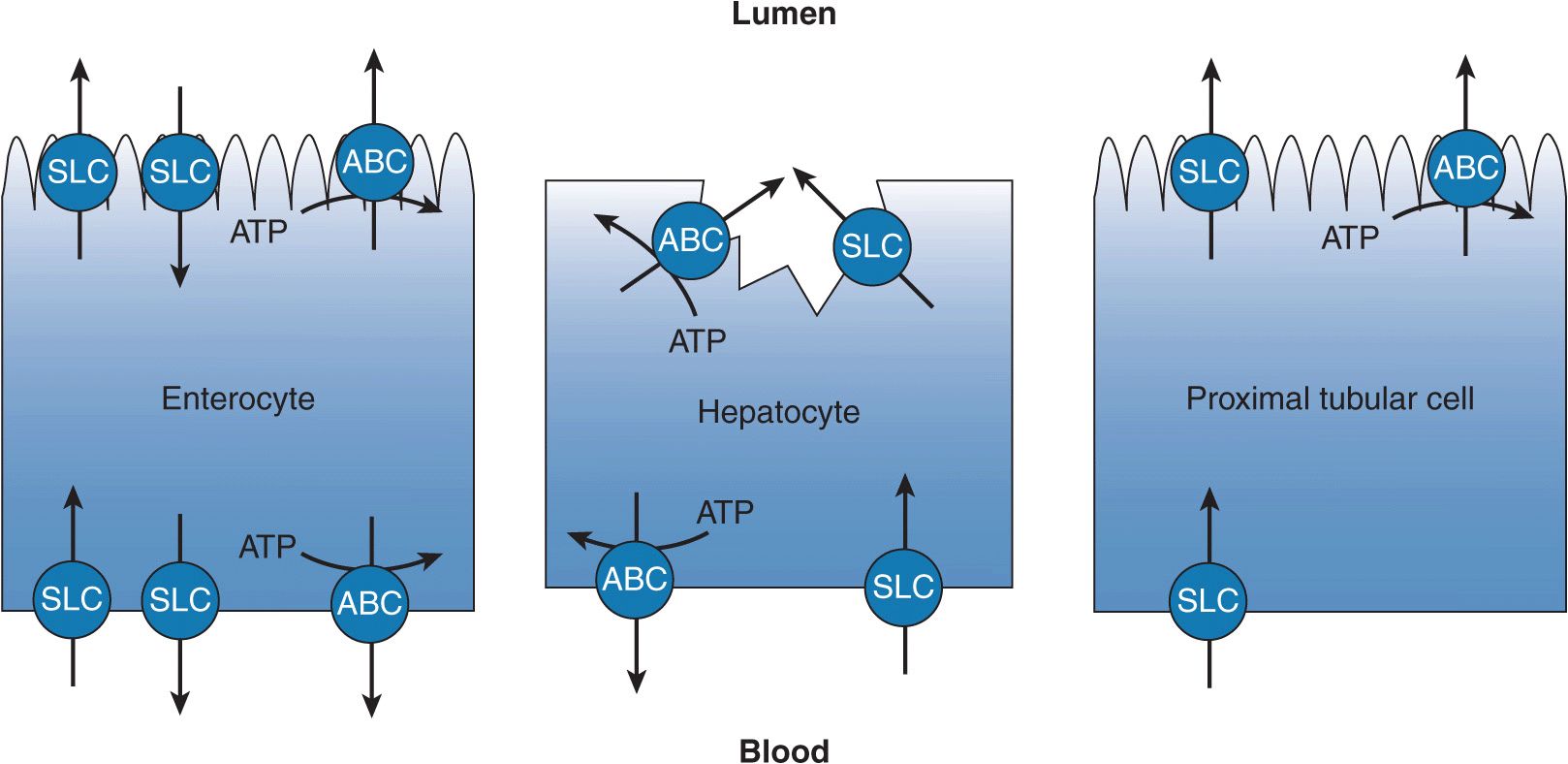
FIGURE 7–1 The transepithelial transport of drugs is facilitated by complementary transporters localized to the two membrane surfaces of polarized epithelia. Illustrated are transport processes across the enterocyte (small intestine), the hepatocyte (liver), and the cells of the renal proximal tubule (kidney). The enterocyte generally is responsible for limiting drug absorption into the blood, whereas the hepatocyte and the cells of the renal proximal tubule facilitate the excretion of drug. Abbreviations: solute carrier (SLC) transporter and ATP-binding cassette (ABC) transporter.
Although transporters have different substrate specificities, there is overlap and redundancy in the types of substrates they transport. For example, P-gp transports a wide variety of substrates that are structurally unrelated, but are frequently either neutral or positively charged hydrophobic compounds such as antiviral agents, immunosuppressive agents, and chemotherapy agents. MRP2 also transports the chemotherapy agents methotrexate and irinotecan. BCRP (ABCG2) mediates the efflux of chemotherapy agents, antiviral drugs, HMG-CoA reductase inhibitors, calcium channel blockers, and steroid metabolites. PEPT1 mediates the absorption of beta-lactam antibiotics, angiotensin-converting enzyme inhibitors, and antiviral drugs.
TRANSPORTERS IN THE LIVER
Drugs are largely cleared by the liver, primarily by metabolic means, and also through transport mechanisms. Hepatic drug elimination through transport mechanisms, which is biliary clearance, is initiated by flux of drug into the hepatocyte across the sinusoidal membrane, by either passive diffusion or transportermediated uptake. Once inside the hepatocyte the drug may or may not be subject to intracellular metabolism by cytochromes P450 and/or conjugation by phase II enzymes. The metabolite, or unchanged drug, is then secreted into the bile across the canalicular membrane or back into the blood across the sinusoidal membrane by transporters that mediate the efflux of drug. The vectorial flux of drug through the hepatocyte is attributable to its polarized nature. The hepatocyte has basolateral (sinusoidal) and apical (canalicular) membranes that differ in transporter composition and consequently function (Figure 7–1). The function of the transporters localized to each membrane may be limited by the kinetic properties of the transporter, affected by the presence of a transporter inhibitor, or diminished by genetic mutations.
Drug Uptake in the Liver
The movement of drugs across the sinusoidal membrane of the hepatocyte is generally facilitated by high concentrations of drug in portal circulation following administration. The high concentrations of drug in the portal circulation create a concentration gradient that favors diffusion into the hepatocyte for drugs that are highly permeable and also favors the transporter-mediated uptake of drugs that are poorly permeable. Hepatic drug transport is primarily characterized by the translocation of large (MW greater than approximately 400 g/mol), somewhat hydrophobic organic anions and amphipathic organic molecules. The uptake of organic anions by the hepatocyte is most frequently associated with three organic anion-transporting polypeptides (OATPs), OATP1B1 (SLCO1B1), OATP1B3 (SLCO1B3), and OATP2B1 (SLCO2B1). The mechanism of organic anion transport by the OATPs is an electroneutral process; however, the associated charge-carrying ion has not been identified. Common substrates of the OATPs include pravastatin, valsartan, glyburide, and repaglinide. Complementing the OATPs is OCT1. It transports a variety of relatively small (less than approximately 400 g/mol), polar cationic molecules across the sinusoidal membrane of the hepatocyte and may facilitate the metabolism of weak bases, such as debrisoquine, by the cytochromes P450. Other substrates of OCT1 include metformin and tropisetron.
Drug Efflux from the Liver
Transporters that mediate drug efflux from the hepatocyte generally belong to the ABC subfamily. ABC transporters are localized to both the sinusoidal membrane and the canalicular membrane where they mediate drug efflux from the hepatocyte into the blood and into the bile, respectively. Transporters localized to the sinusoidal membrane include MRP3 (ABCC3), MRP4 (ABCC4), and MRP6 (ABCC6); these transporters mediate drug or drug-conjugate efflux from the hepatocyte. The transport of phase II drug conjugates into the blood by MRP3, 4, or 6 may promote the renal clearance of the more polar drug conjugates, supplementing any biliary clearance by canalicular membrane efflux transporters. ABC transporters localized to the canalicular membrane include P-gp, MRP2 (ABCC2), BCRP, and BSEP. These transporters facilitate the transport of drugs and endogenous molecules and are frequently associated with the biliary clearance of drugs. Canalicular efflux transporters demonstrate a broad range of substrate specificity that includes endogenous and exogenous molecules. Molecules transported by the canalicular ABC transporters are generally moderately lipophilic, usually >400 g/mol, and are typically differentiated by charge. For example, P-gp commonly transports cationic molecules (i.e., weak bases) such as loperamide, fexofenadine, and vinblastine, while MRP2 and BCRP frequently transport anionic compounds, including phase II conjugates, such as valsartan, methotrexate, pitavastatin, and estrone-3-sulfate. While the substrates of the canilicular transporters are often differentiated by charge, uncharged molecules, or molecules having an atypical charge, may also be transported (e.g., digoxin is transported by P-gp and prazosin is transported by BCRP). In addition to the ABC transporters, the SLC transporter, MATE1, is also expressed on the canalicular membrane and transports relatively small organic cations in exchange for a proton.
TRANSPORTERS IN THE KIDNEY
For drugs having limited or negligible metabolic or biliary clearance, the excretion of unchanged drug by the kidneys can represent a significant route of drug elimination. Additionally, renal clearance is a principal means of clearing metabolites of drugs and drug conjugates. The transepithelial translocation (secretion) of drugs from the interstitial space to the tubule lumen is a two-step process (Figure 7–1) and is additive to elimination by glomerular filtration. The proximal tubule is the primary site within the nephron for the secretion of drugs. The first step of the sequential process is the transport of drug across the epithelial cell’s basolateral membrane into the cytoplasm. The second step is the transport of drug across the cell’s apical membrane by a transporter whose substrate specificity is complementary to the basolateral transporter facilitating the first step in the process. Simple diffusion may substitute for one of the steps provided a drug has sufficient passive permeability. The activity of these drug transporters is characterized by Michaelis–Menten kinetics, therefore the secretory process is saturable and may be inhibited by coadministrated drugs. Furthermore, the kinetic activity of these transporters may be increased or, more frequently, decreased by changes in the amino acid sequence of the protein, changes that are subsequent to changes in the genome. The transepithelial secretion of drugs therefore can be saturated, inhibited, and is subject to genetic mutations (single nucleotide polymorphisms [SNPs]). Thus, the renal clearance of drugs by transporters may be limited at high drug concentrations, restricted by coadministered drugs, or diminished by gene mutations that reduce protein function or expression.
Organic Cation Transport in Kidney
The mechanism of organic cation secretion most likely has evolutionary precedent for clearing the body of xenobiotics, such as plant alkaloids, largely acquired through dietary means, or environmental toxins, such as nicotine. However, in the modern era it plays an important role in the elimination of therapeutic drugs. As such, the secretory process is subject to clinically significant interactions with drugs. For example, the substrates metformin and procainamide show reduced renal clearances in the presence of the transporter inhibitor cimetidine. Common substrates of the processes surrounding organic cation transport in the kidney include metformin, varenicline, procainamide, and amiloride. These compounds reflect the physicochemical properties of the process, that is, they are generally ≤400 g/mol, relatively polar, and weak bases.
The secretion of organic cations is a two-step process. The first step, entry into the cell across the basolateral membrane of proximal tubule cells, is a uniport (facilitated diffusion) mechanism, driven by an electrical and chemical gradient for the substrate, and is mediated by the OCT2. The electrical component is derived from the negative membrane potential and the chemical gradient reflects the concentration gradient. An alternative means for cellular entry across the basolateral membrane is by simple diffusion. This also represents an electrically conductive means for entry and more commonly characterizes the entry step for hydrophobic organic cations.
The complementary exit step of organic cations across the luminal (apical) membrane involves an active transport process of carrier-mediated exchange (antiport) of an intracellular organic cation for one extracellular hydrogen ion (H+), or proton, and is mediated by the multidrug and toxin extrusion family members MATE1 and MATE2-K. The electroneutral antiport is driven by the proton (pH) gradient, because the proximal tubule lumen is acidic relative to that of the cells lining the lumen.
The coordinated actions of the basolateral transport process, uniport (facilitated diffusion), and the apical transport process, antiport, result in net transepithelial secretion of organic cations. An alternative means of apical secretion of organic cations is by an ATP-dependent process, where ATP provides the energy for a mechanism of primary active transport of molecules across the apical (luminal) membrane. This mechanism of secretory flux is more frequently associated with large-molecular-weight organic cations (>400 g/mol), and is facilitated by the apically localized P-gp.
The cellular processes associated with secretion of organic cations can be summarized as follows: (1) low-molecular-weight organic cations enter proximal cells across the basolateral membrane via electrogenic process of facilitated diffusion and exit cells across the luminal/apical membrane by means of electroneutral exchange for H+; (2) larger-molecular-weight organic cations likely diffuse into proximal tubule cells, or may be transported by an as yet to be defined transporter, across the peritubular membrane and are pumped across the apical membrane into the tubular lumen by the primary active transporter, P-gp. Notably, overlap appears to exist in the transport pathways for small and large organic cations. Furthermore, the transepithelial transport of organic cations in renal proximal tubules involves the parallel activity of multiple discrete transport processes.
Organic Anion Transport in the Kidney
The mechanism of organic anion secretion likely parallels that of organic cation transport in that it has evolutionary precedent for clearing the body of xenobiotics. However, much like the organic cation transporters, organic anion transporters play an important role in the elimination of therapeutic drugs. As such, the secretory process is subject to clinically significant interactions with drugs. For example, the clearance of adefovir and furosemide is inhibited by the presence of probenecid. Common substrates of the processes surrounding organic anion transport in the kidney include adefovir, penicillin G, and methotrexate. Additionally, substrates of organic anion transport processes frequently include a number of products of hepatic biotransformation. These compounds reflect the physicochemical properties of the process, that is, they are generally ≤400 g/mol, relatively polar, and are weak acids.
The secretion of organic anions is also a two-step process. The coordinated actions of the basolateral transport process, anion exchange, and the apical transport processes, facilitated diffusion and active transport, result in net transepithelial secretion of organic anions. The first step, entry into the cell across the basolateral membrane of proximal tubule cells, is an active step characterized as an exchange (antiport) mechanism, where intracellular dicarboxylate ions (e.g., α-ketoglutarate, a Kreb’s cycle intermediate) are exchanged for organic anions. The antiporters responsible for mediating the cellular uptake of organic anions into proximal tubule cells are the organic anion transporters OAT1 and OAT3. An alternative means for cellular entry across the basolateral membrane is by simple diffusion.
The complementary exit step of organic anions across the luminal (apical) membrane has largely been characterized as a transport process of facilitated diffusion. The process of facilitated diffusion is driven by an electrochemical and concentration gradient of the substrate inside the cell driven by the active step at the basolateral membrane. The negative membrane potential provides an electromotive force for the exit of negatively charged compounds, such as organic anions. An alternative means for exit across the apical membrane is by simple diffusion.
Organic anions, similar to organic cations, may also be secreted into the proximal tubule lumen by an ATP-dependent process, where ATP provides the energy for a mechanism of primary active transport. However, rather than being mediated by P-gp, the primary active transport of organic anions across the luminal membrane is commonly facilitated by multidrug resistance proteins, MRP2 and MRP4.
The cellular processes associated with secretion of organic anions can be summarized as follows: (1) low-molecular-weight organic cations enter proximal cells across the basolateral membrane in exchange for intracellular dicarboxylates (e.g., α-ketoglutarate) and exit cells across the luminal/apical membrane by facilitated diffusion; (2) alternatively, organic anions may also enter by the same mechanism, but may be pumped across the apical membrane into the tubular lumen by the primary active transporter, such as MRP2 or MRP4. Notably, overlap exists in the two transport pathways.
TRANSPORTERS INVOLVED IN THE BLOOD–BRAIN BARRIER
The BBB is an interface between the brain and the systemic circulation within the capillary endothelium and controls what goes in and out of the central nervous system (CNS). This barrier is known to be highly active, dynamic, and selective in mediating signals coming from the periphery and brain. The barrier function of the BBB consists of three components: tight junctions, metabolizing enzymes, and transporters. Transporters are an important component of the BBB that function as active barriers to protect the brain from drugs and other neuroactive and toxic substances. Transporters from both the SLC and ABC superfamilies are involved in the BBB. Transporters from the SLC superfamily facilitate influx of glucose, amino acids, ions, and other nutrients needed to meet the energy demands of the brain. Efflux transporters from the ABC superfamily actively clear wastes and prevent xenobiotics and other toxins from entering the brain. While the BBB transporters are an important barrier to the permeation of unwanted substances into the brain, transporters can also be an impediment when trying to get therapeutic substances into the CNS. For example, the presence of ABC transporters may limit penetration of chemotherapeutics into the CNS creating a safe harbor for brain cancers to thrive.
P-gp, BCRP, and MRP transporters are involved in limiting drug entry into BBB. P-gp is the most extensively studied with the most established examples of limiting drug penetration into the brain. The role of P-gp at the BBB was demonstrated in a study of healthy volunteers. Coadministration of the antidiarrheal agent and potent opioid, loperamide, and the P-gp inhibitor quinidine caused significant respiratory depression, a CNS side effect of opioid administration. By contrast, when loperamide was administered alone no respiratory effect was observed, because P-gp acts to prevent CNS penetration of loperamide.3 For this reason, loperamide, while it is a potent opioid, is available over-the-counter as an antidiarrheal medication. P-gp and BCRP have overlapping substrate specificities, and are thought to cooperate in limiting drug entry into the brain. Several isoforms of MRP have been identified at the BBB: MRP1, MRP2, MRP3, MRP4, and MRP5. MRPs transport organic anions, glutathione, glucuronide- or sulfate-conjugated compounds, and nucleoside analogs.
P-GLYCOPROTEIN: ABCB1
P-gp, encoded by the gene ABCB1 (formerly known as MDR1), is a 1,280 amino acid transmembrane protein that is important in the efflux of a wide variety of drugs and endogenous compounds. In addition to the tissues previously described, P-gp is expressed at various blood–tissue barriers such as the blood–brain, blood–cerebrospinal fluid (CSF), blood–testis, and blood–placenta barriers, functioning to limit entry of xenobiotic substances into these tissues. Consistent with its ability to limit the accumulation of xenobiotics in tissues, P-gp was the first transporter identified as a facilitator of tumor cell drug resistance (multidrug resistance) by mediating efflux of chemotherapeutics from tumor cells. The role P-gp has in limiting the intestinal absorption of compounds, promoting the excretion of compounds by the liver and kidney, and limiting tissue accumulation illustrates its responsibility in the pharmacokinetics of substrate compounds.
P-gp demonstrates broad substrate diversity; P-pg substrates include analgesics, anticancer agents, HIV and HCV protease inhibitors, immunosuppressives, corticosteroids, and antibiotics (see Table 7–1). Inhibitors and inducers of P-gp are equally diverse. Clinically this diversity is important to the underlying mechanism of drug–drug interactions. Determining drug–drug interactions involving P-gp substrates is not straightforward because P-gp has multiple drug-binding sites with different binding affinities. Therefore, determining drug interactions with P-gp substrates may require multiple drug pairs to cover the range of substrates that bind to the different binding sites.4 Additionally, multiple substrates may be required to assess the effect of a given polymorphism if the binding pocket is altered in a way that impacts some compounds but not others. There are currently no recommendations to address the potential for multiple P-gp-binding sites.2
TABLE 7–1 Transporters and representative substrates.
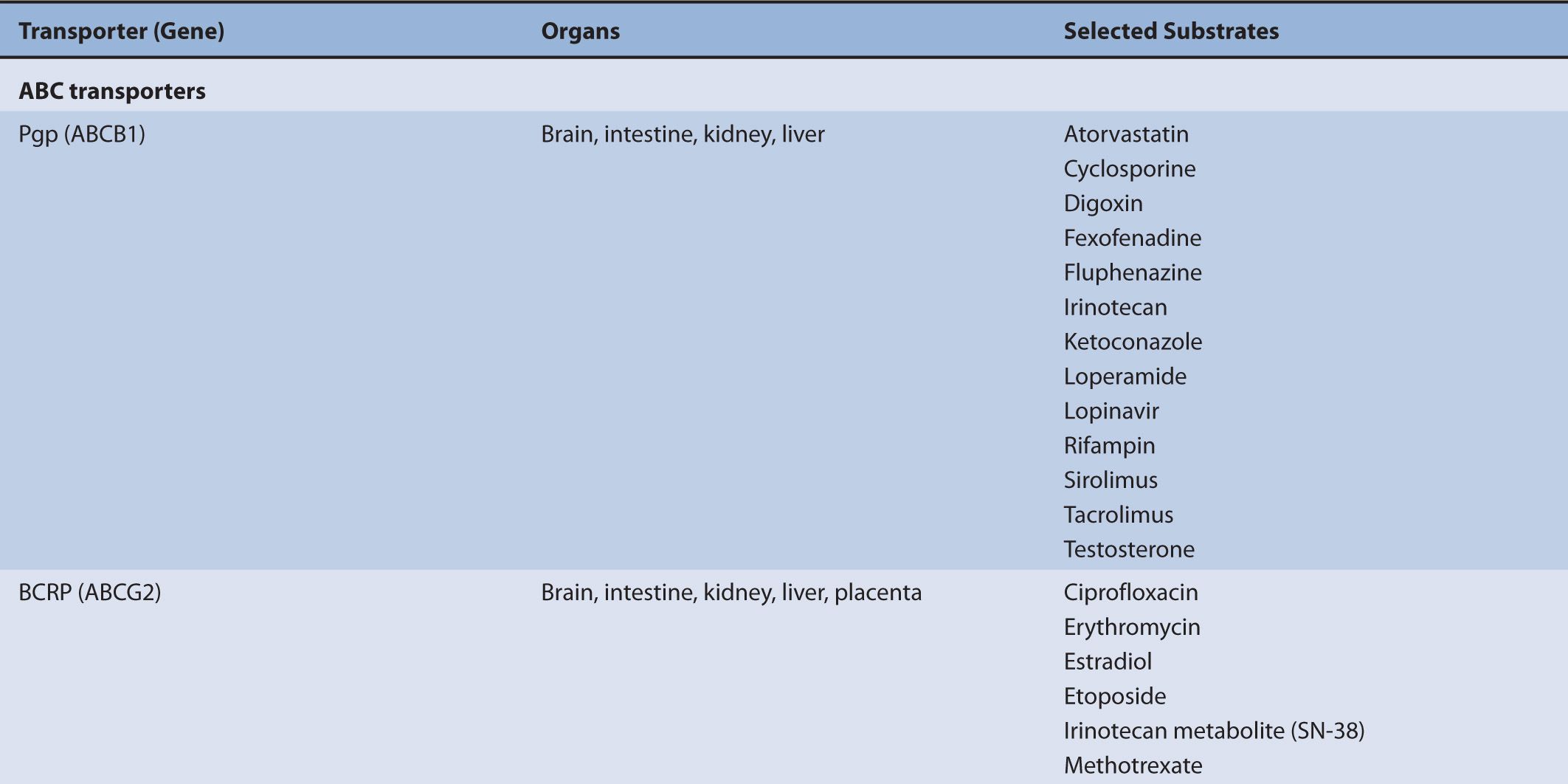
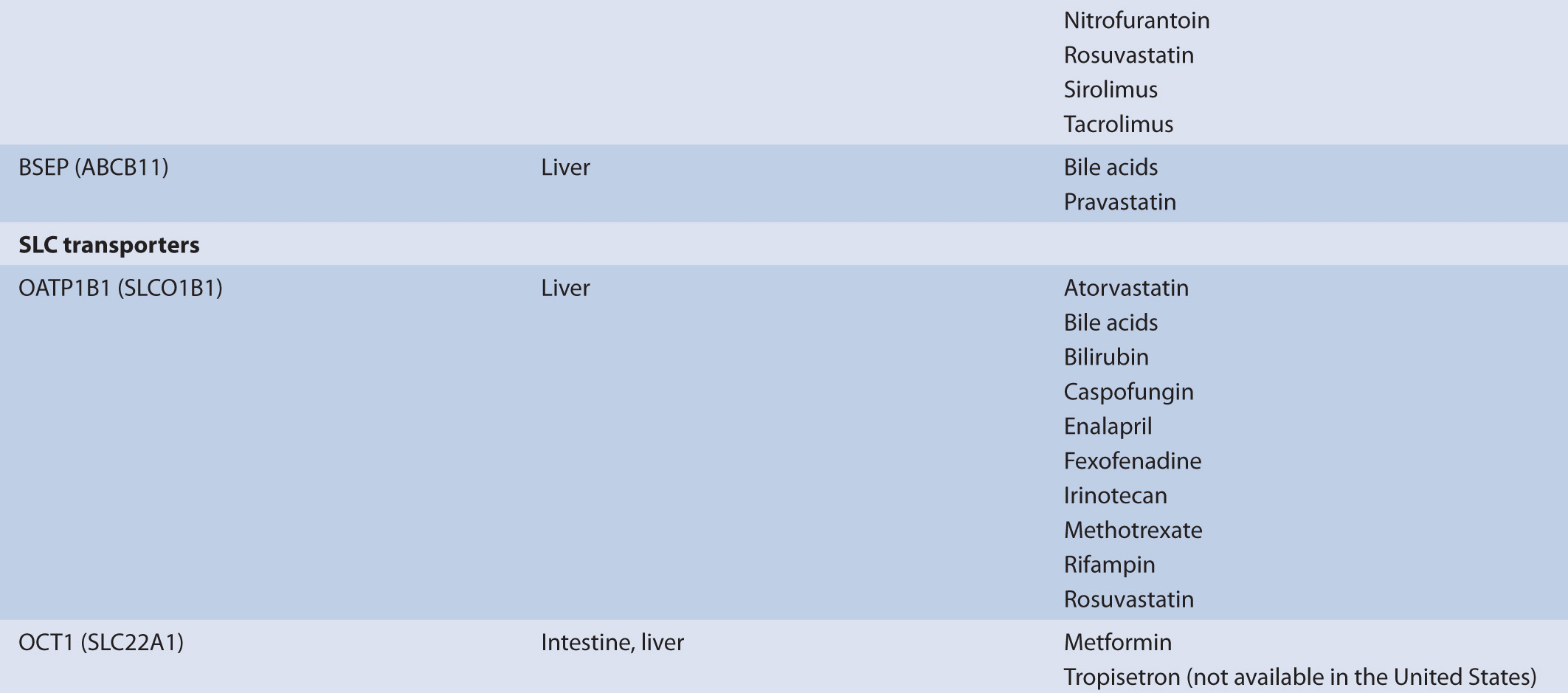
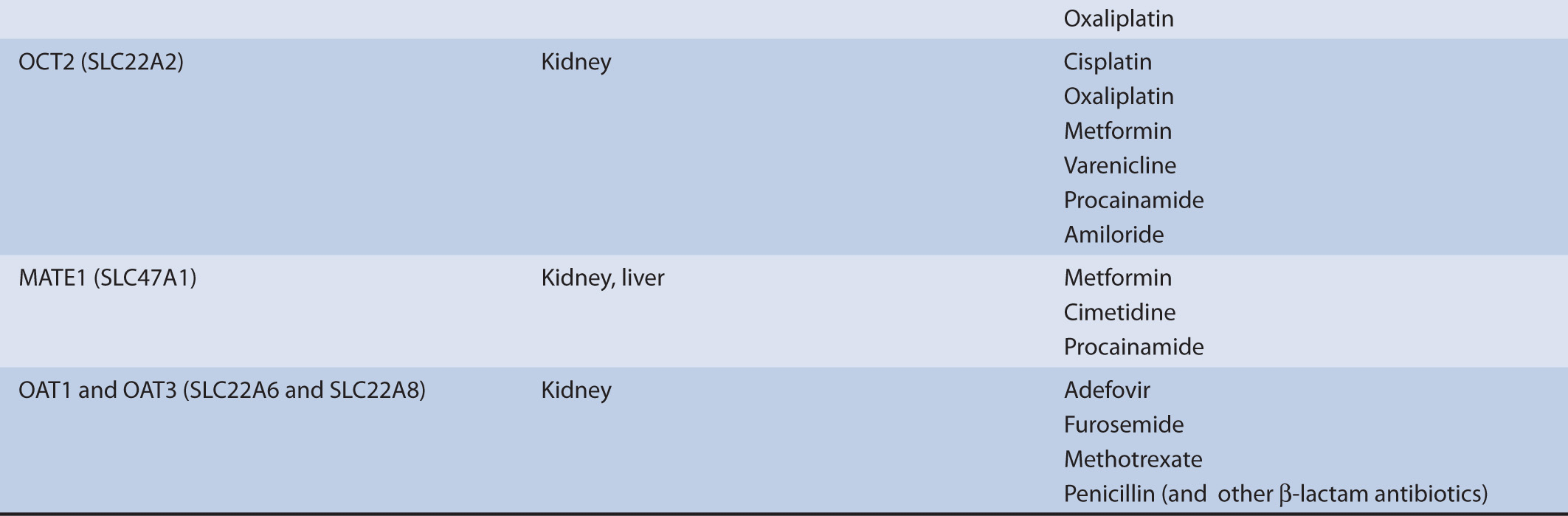
P-gp may also complement other transporters and drug-metabolizing enzymes in the intestine, liver, and kidney to affect drug concentrations in the tissue and plasma. For example, both CYP3A4 and P-gp are highly expressed in the intestine and liver and have broad overlapping substrate specificity. One hypothesis suggests that CYP3A4 and P-gp complement each other by limiting oral drug bioavailability.5 The presence of P-gp allows a drug molecule more opportunity for CYP3A metabolism in the intestine, thus decreasing overall systemic exposure. Through repeated cycles of passive absorption and subsequent efflux by P-gp, there is an increased residence time of the drug in the enterocyte providing more opportunity for intestinal metabolism by CYP3A. Additionally, this coordinated action may play a role in drug interactions and adverse drug effects. Therefore, in order to accurately assess the mechanism of a potential drug interaction for a given P-gp substrate, the contributions from intestinal and hepatic P-gp, CYP3A4, and other transporters may need to be considered.
In addition to the diminished P-gp activity caused by drug–drug interactions, P-gp activity can be increased or decreased by a number of other factors. The expression of P-gp, and correspondingly its activity, can be inhibited or induced by drugs, disease states, or genetic polymorphisms. Many investigative studies have shown alterations in P-gp activity due to genetic polymorphisms or drug–drug interactions; however, whether these alterations are clinically significant remains unknown.
As discussed in other chapters, SNPs are one of the most important mechanisms of genetic variability. There are at least 1,200 SNPs in the ABCB1 gene with approximately 100 in the coding region.6 Most of the human studies have focused on the three most common SNPs—rs1128503 (1236C>T, Gly412Gly), resulting in a synonymous change; rs2032582 (2677G>T/A, Ser893Ala/Thr), a nonsynonymous (or silent) SNP; and rs1045642 (3435C>T, Ile1145Ile), a synonymous SNP—which are in high linkage disequilibrium. There are thought to be four important haplotypes associated with these SNPs: 1236C/2677G/3435C (ABCB1*1), which is the reference sequence; 1236T/2677T/3435T (ABCB1*2); 1236C/2677G/3435T (also named ABCB1*2); and 1236T/2677T/3435T plus three intronic SNPs (ABCB1*13). The allelic frequency of the 3435C>T SNP varies with ethnicity with the 3435T variant allele occurring in greater frequency in Caucasians (Caucasians; 0.52–0.57) compared with Asians (Asians; 0.41–0.47) or African blacks (0.17–0.27).6
The functional activity of P-gp is dependent on two major factors: (1) the gene expression of ABCB1 that controls the amount of protein available and (2) the functionality of P-gp that determines which substrates will bind and how efficiently they will bind. Amino acid changes within the P-gp protein can influence this functionality. Interestingly, one of the common ABCB1 SNPs, ![]() has been shown to affect P-gp transport function in vitro even though the amino acid sequence was unchanged. This so-called silent polymorphism is thought to alter the conformation of the substrate and inhibitor interaction site, thus altering the ability of P-gp to bind to a substrate.7
has been shown to affect P-gp transport function in vitro even though the amino acid sequence was unchanged. This so-called silent polymorphism is thought to alter the conformation of the substrate and inhibitor interaction site, thus altering the ability of P-gp to bind to a substrate.7
Effect of ABCB1 Genetic Polymorphisms on Drug Pharmacokinetics
In order to determine if genetic variability in the ABCB1 gene causes changes in P-gp activity, clinical studies look for changes in the pharmacokinetics of representative drugs known to be transported primarily by P-gp. P-gp affects pharmacokinetics through its role in limiting oral absorption that can be measured by area under the concentration-versus-time curve (AUC) or maximum concentration (Cmax). For example, if a genetic polymorphism in the ABCB1 gene causes increased P-gp expression, one would expect decreased oral bioavailability, decreased AUC, decreased Cmax, and increased renal clearance (exemplified as apical transporter in Figure 7–2a to 7–2c). In addition, studies can investigate changes in target cell or organ tissue concentrations in which P-gp limits entry of drugs.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


