Drug Therapy for Hypercholesterolemia and Dyslipidemia
Hyperlipidemia is a major cause of atherosclerosis and atherosclerosis-induced conditions, such as coronary heart disease (CHD), ischemic cerebrovascular disease, and peripheral vascular disease. These conditions cause morbidity or mortality in a majority of middle-aged or older adults. Dyslipidemias, including hyperlipidemia (hypercholesterolemia) and low levels of high-density-lipoprotein cholesterol (HDL-C), are major causes of increased atherogenic risk; both genetic disorders and lifestyle contribute to the dyslipidemias seen in countries around the world.
Classes of drugs that modify cholesterol levels include:
• Statins, inhibitors of 3-hydroxy-3-methylglutaryl–coenzyme A (HMG-CoA) reductase
• Bile acid–binding resins
• Nicotinic acid (niacin)
• Fibric acid derivatives
• Ezetimibe, an inhibitor of cholesterol absorption
These drugs provide benefit in patients across the entire spectrum of cholesterol levels, primarily by reducing levels of low-density-lipoprotein cholesterol (LDL-C). Drug regimens that reduce LDL-C levels moderately (30-40%) can reduce fatal and nonfatal CHD events and strokes by as much as 30-40%. In patients with low HDL-C and average LDL-C levels, appropriate drug therapy reduces CHD endpoint events by 20-35%. Because two-thirds of patients with CHD in the U.S. have low HDL-C levels (<40 mg/dL in men, <50 mg/dL in women), low-HDL-C patients should be treated for dyslipidemia, even if their LDL-C levels are in the normal range.
Severe hypertriglyceridemia (i.e., triglyceride levels of >1000 mg/dL) requires therapy to prevent pancreatitis. Moderately elevated triglyceride levels (150-400 mg/dL) are of concern because they often occur as part of the metabolic syndrome, which includes insulin resistance, obesity, hypertension, low HDL-C levels, a procoagulant state, and substantially increased risk of CVD. Atherogenic dyslipidemia in patients with metabolic syndrome also is characterized by lipid-depleted LDL (sometimes referred to as “small, dense LDL”). Metabolic syndrome affects ~25% of adults and is common in CVD patients; hence, identification of moderate hypertriglyceridemia in a patient, even if the total cholesterol level is normal, should trigger an evaluation to identify insulin-resistant patients with this disorder.
PLASMA LIPOPROTEIN METABOLISM
Lipoproteins are macromolecular assemblies that contain lipids and proteins. The lipid constituents include free and esterified cholesterol, triglycerides, and phospholipids. The protein components, known as apolipoproteins or apoproteins, provide structural stability to the lipoproteins and also may function as ligands in lipoprotein–receptor interactions or as cofactors in enzymatic processes that regulate lipoprotein metabolism. The major classes of lipoproteins and their properties are summarized in Table 31–1. Apoproteins have well-defined roles in plasma lipoprotein metabolism (Table 31–2).
Table 31–1
Characteristics of Plasma Lipoproteins
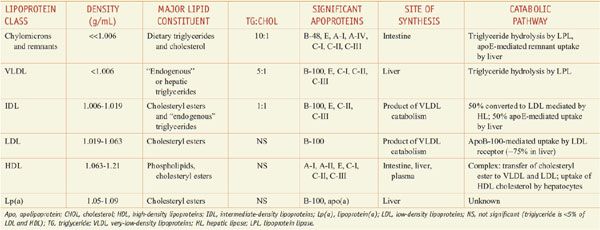
Table 31–2
Apolipoproteins
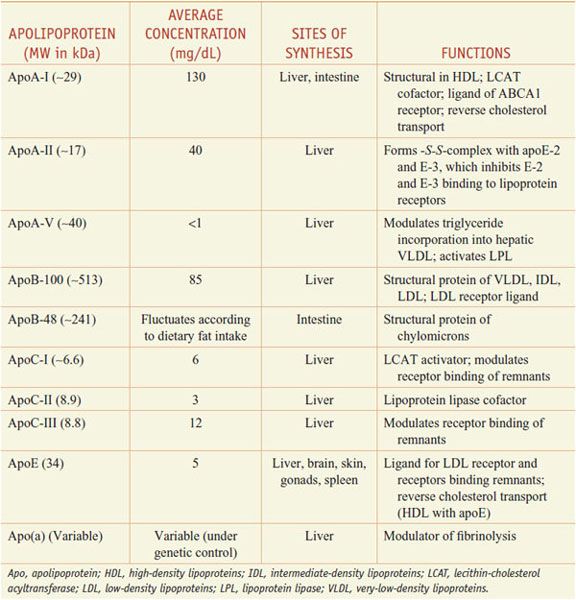
In all spherical lipoproteins, the most water-insoluble lipids (cholesteryl esters and triglycerides) are core components, and the more polar, water-soluble components (apoproteins, phospholipids, and unesterified cholesterol) are located on the surface. Except for apo(a), the lipid-binding regions of all apoproteins contain amphipathic helices that interact with the polar, hydrophilic lipids (such as surface phospholipids) and with the aqueous plasma environment in which the lipoproteins circulate. Differences in the non–lipid-binding regions determine the functional specificities of the apolipoproteins.
Figure 31–1 summarizes the pathways involved in the uptake and transport of dietary fat and cholesterol, pathways that involve the lipoprotein structures described below.
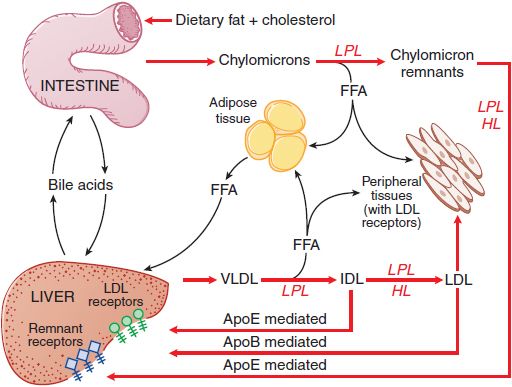
Figure 31–1 The major pathways involved in the metabolism of chylomicrons synthesized by the intestine and VLDL synthesized by the liver. Chylomicrons are converted to chylomicron remnants by the hydrolysis of their triglycerides by LPL. Chylomicron remnants are rapidly cleared from the plasma by the liver. “Remnant receptors” include the LDL receptor–related protein (LRP), LDL receptors, and perhaps other receptors. Free fatty acid (FFA) released by LPL is used by muscle tissue as an energy source or taken up and stored by adipose tissue. HL, hepatic lipase; IDL, intermediate-density lipoproteins; LDL, low-density lipoproteins; LPL, lipoprotein lipase; VLDL, very-low-density lipoproteins.
CHYLOMICRONS. Chylomicrons are synthesized from the fatty acids of dietary triglycerides and cholesterol absorbed by epithelial cells in the small intestine. Chylomicrons are the largest and lowest density plasma lipoproteins. In normolipidemic individuals, chylomicrons are present in plasma for 3-6 h after a fat-containing meal has been ingested. Intestinal cholesterol absorption is mediated by Niemann-Pick C1–Like 1 protein (NPC1L1), which appears to be the target of ezetimibe, a cholesterol absorption inhibitor.
After their synthesis in the endoplasmic reticulum, triglycerides are transferred by microsomal triglyceride transfer protein (MTP) to the site where newly synthesized apoB-48 is available to form chylomicrons. ApoB-48, synthesized only by intestinal epithelial cells, is unique to chylomicrons and functions primarily as a structural component of chylomicrons. Dietary cholesterol is esterified by the type 2 isozyme of acyl coenzyme A:cholesterol acyltransferase (ACAT-2). ACAT-2 is found in the intestine and in the liver, where cellular free cholesterol is esterified before triglyceride-rich lipoproteins (chylomicrons and very-low-density lipoproteins [VLDL]) are assembled.
After entering the circulation via the thoracic duct, chylomicrons are metabolized initially at the capillary luminal surface of tissues that synthesize lipoprotein lipase (LPL), (see Figure 31–1), including adipose tissue, skeletal and cardiac muscle, and breast tissue of lactating women. The resulting free fatty acids are taken up and used by the adjacent tissues. The interaction of chylomicrons and LPL requires apoC-II as a cofactor.
CHYLOMICRON REMNANTS. After LPL-mediated removal of much of the dietary triglycerides, the chylomicron remnants, with all of the dietary cholesterol, detach from the capillary surface and within minutes are removed from the circulation by the liver (see Figure 31–1). First, the remnants are sequestered by the interaction of apoE with heparan sulfate proteoglycans on the surface of hepatocytes and are processed by hepatic lipase (HL), further reducing the remnant triglyceride content. Next, apoE mediates remnant uptake by interacting with the hepatic LDL receptor or the LDL receptor–related protein (LRP).
During the initial hydrolysis of chylomicron triglycerides by LPL, apoA-I and phospholipids are shed from the surface of chylomicrons and remain in the plasma. This is one mechanism by which nascent (precursor) HDL are generated. Chylomicron remnants are not precursors of LDL, but the dietary cholesterol delivered to the liver by remnants increases plasma LDL levels by reducing LDL receptor–mediated catabolism of LDL by the liver.
VERY-LOW-DENSITY LIPOPROTEINS. VLDL are produced in the liver when triglyceride production is stimulated by an increased flux of free fatty acids or by increased de novo synthesis of fatty acids by the liver.
ApoB-100, apoE, and apoC-I, C-II, and C-III are synthesized constitutively by the liver and incorporated into VLDL (see Table 31–2). Triglycerides are synthesized in the endoplasmic reticulum, and along with other lipid constituents, are transferred by MTP to the site in the endoplasmic reticulum where newly synthesized apoB-100 is available to form nascent (precursor) VLDL. Small amounts of apoE and the C apoproteins are incorporated into nascent particles within the liver before secretion, but most of these apoproteins are acquired from plasma HDL after the VLDL are secreted by the liver. Mutations of MTP that result in the inability of triglycerides to be transferred to either apoB-100 in the liver or apoB-48 in the intestine prevent VLDL and chylomicron production and cause the genetic disorder abetalipoproteinemia.
Plasma VLDL is catabolized by LPL in the capillary beds in a process similar to the lipolytic processing of chylomicrons (see Figure 31–1). When triglyceride hydrolysis is nearly complete, the VLDL remnants, usually termed IDL, are released from the capillary endothelium and reenter the circulation. ApoB-100 containing small VLDL and IDL, which have a t1/2 <30 min, have 2 potential fates. About 40-60% are cleared from the plasma by the liver via apoB-100 and apoE-mediated interaction with LDL receptors and LRP. LPL and HL convert the remainder of the IDL to LDL by removal of additional triglycerides. The C apoproteins, apoE, and apoA-V redistribute to HDL.
ApoE plays a major role in the metabolism of triglyceride-rich lipoproteins (chylomicrons, chylomicron remnants, VLDL, and IDL). About half of the apoE in the plasma of fasting subjects is associated with triglyceride-rich lipoproteins, and the other half is a constituent of HDL.
LOW-DENSITY LIPOPROTEINS. Virtually all of the LDL particles in the circulation are derived from VLDL. The LDL particles have a t1/2 of 1.5-2 days. In subjects without hypertriglyceridemia, two-thirds of plasma cholesterol is found in the LDL. Plasma clearance of LDL is mediated primarily by LDL receptors (ApoB-100 binds LDL to its receptor); a small component is mediated by nonreceptor clearance mechanisms.
The most common cause of autosomal dominant hypercholesterolemia involves mutations of the LDL receptor gene. Defective or absent LDL receptors cause high levels of plasma LDL and familial hypercholesterolemia. LDL becomes atherogenic when modified by oxidation, a required step for LDL uptake by the scavenger receptors of macrophages. This process leads to foam-cell formation in arterial lesions. At least 2 scavenger receptors (SRs) are involved (SR-AI/II and CD36). SR-AI/II appears to be expressed more in early atherogenesis, and CD36 expression is greater as foam cells form during lesion progression. The liver expresses a large complement of LDL receptors and removes ~75% of all LDL from the plasma. Consequently, manipulation of hepatic LDL receptor gene expression is a most effective way to modulate plasma LDL-C levels. Thyroxine and estrogen enhance LDL receptor gene expression, which explains their LDL-C–lowering effects. The most effective dietary alteration (decreased consumption of saturated fat and cholesterol) and pharmacological treatment (statins) for hypercholesterolemia act by enhancing hepatic LDL receptor expression.
HIGH-DENSITY LIPOPROTEINS. HDL are protective lipoproteins that decrease the risk of CHD; thus, high levels of HDL are desirable. This protective effect may result from the participation of HDL in reverse cholesterol transport, the process by which excess cholesterol is acquired from cells and transferred to the liver for excretion. HDL effects also include putative anti-inflammatory, anti-oxidative, platelet anti-aggregatory, anticoagulant, and profibrinolytic activities. ApoA-I is the major HDL apoprotein, and its plasma concentration is a more powerful inverse predictor of CHD risk than is the HDL-C level. ApoA-I synthesis is required for normal production of HDL.
Mutations in the apoA-I gene that cause HDL deficiency often are associated with accelerated atherogenesis. In addition, 2 major subclasses of mature HDL particles in the plasma can be differentiated by their content of the major HDL apoproteins, apoA-I and apoA-II. Epidemiologic evidence in humans suggests that apoA-II may be atheroprotective.
The membrane transporter ABCA1 facilitates the transfer of free cholesterol from cells to HDL. After free cholesterol is acquired by the pre-β1 HDL, it is esterified by lecithin:cholesterol acyltransferase. The newly esterified and nonpolar cholesterol moves into the core of the particle, which becomes progressively more spherical, larger, and less dense with continued cholesterol acquisition and esterification. As the cholesteryl ester content of the particle (now called HDL2) increases, the cholesteryl esters of these particles begin to be exchanged for triglycerides derived from any of the triglyceride-containing lipoproteins (chylomicrons, VLDL, remnant lipoproteins, and LDL). This exchange, mediated by the cholesteryl ester transfer protein (CETP), accounts for the removal of about two-thirds of the cholesterol associated with HDL in humans. The transferred cholesterol subsequently is metabolized as part of the lipoprotein into which it was transferred.
Treatments that target CETP and the ABC transporters have yielded equivocal results in humans. While CETP inhibitors effectively reduce LDL, they also paradoxically increase the frequency of adverse cardiovascular events (angina, revascularization, myocardial infarction, heart failure, and death).
The triglyceride that is transferred into HDL2 is hydrolyzed in the liver by HL, a process that regenerates smaller, spherical HDL3 particles that recirculate and acquire additional free cholesterol from tissues containing excess free cholesterol.
HL activity is regulated and modulates HDL-C levels. Androgens increase HL gene expression/activity, which accounts for the lower HDL-C values observed in men than in women. Estrogens reduce HL activity, but their impact on HDL-C levels in women is substantially less than that of androgens on HDL-C levels in men. HL appears to have a pivotal role in regulating HDL-C levels, as HL activity is increased in many patients with low HDL-C levels.
LIPOPROTEIN (a). Lipoprotein(a) [Lp(a)] is composed of an LDL particle that has a second apoprotein, apo(a), in addition to apoB-100. Apo(a) of Lp(a) is structurally related to plasminogen and appears to be atherogenic.
HYPERLIPIDEMIA AND ATHEROSCLEROSIS
The major conventional risk factors for CVD are elevated LDL-C, reduced HDL-C, cigarette smoking, hypertension, type 2 diabetes mellitus, advancing age, and a family history of premature CHD events (men <55 years; women <65 years) in a first-degree relative (Table 31–3). When total cholesterol levels are below 160 mg/dL, CHD risk is markedly attenuated, even in the presence of additional risk factors. This pivotal role of hypercholesterolemia in atherogenesis gave rise to the almost universally accepted cholesterol-diet-CHD hypothesis: elevated plasma cholesterol levels cause CHD, diets rich in saturated (animal) fat and cholesterol raise cholesterol levels, and lowering cholesterol levels reduces CHD risk.
Table 31–3
Risk Factors for Coronary Heart Disease

Modest reductions in total cholesterol and LDL-C are associated with reductions in fatal and nonfatal CHD events but not total mortality. Patients benefit regardless of gender, age, baseline lipid values, or whether they have a prior history of vascular disease or type 2 diabetes mellitus. Statin therapy is effective in preventing first and subsequent atherothrombotic strokes. Moderate doses of statins that lower LDL-C levels by about 40% reduce cardiovascular events by about one-third. More intensive regimens that lower LDL-C by 45-50% reduce CVD events by as much as 50%.
MANAGING PATIENTS WITH DYSLIPIDEMIA: NATIONAL CHOLESTEROL EDUCATION PROGRAM (NCEP) GUIDELINES
Primary prevention involves management of risk factors to prevent a first-ever CHD event. Secondary prevention is aimed at patients who have had a prior CHD event and whose risk factors must be treated aggressively. Recently, the concept of primordial prevention has been applied to CHD. This is a population-based approach to prevention (rather than treatment) that targets smoking, weight management, physical activity, healthy eating habits, cholesterol and glucose levels, and blood pressure.
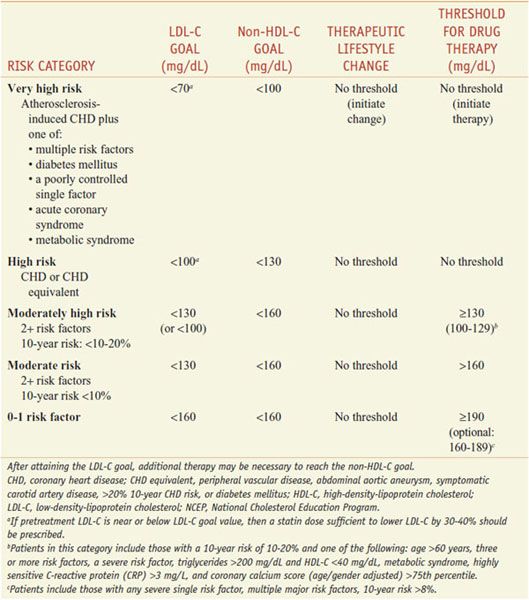
The patient-based approach to manage dyslipidemia is designed for primary and secondary prevention, requires a risk assessment, and focuses on lowering LDL-C and non-HDL-C (see ranges in Table 31–4). Before drug therapy is initiated, secondary causes of hyperlipidemia (Table 31–5) should be excluded. Treatment of the disorder causing secondary dyslipidemia may preclude the necessity of treatment with hypolipidemic drugs. Table 31–6 summarizes current risk category-treatment guidelines based on LDL-C levels.
Table 31–4
Classification of Plasma Lipid Levels (mg/dL)
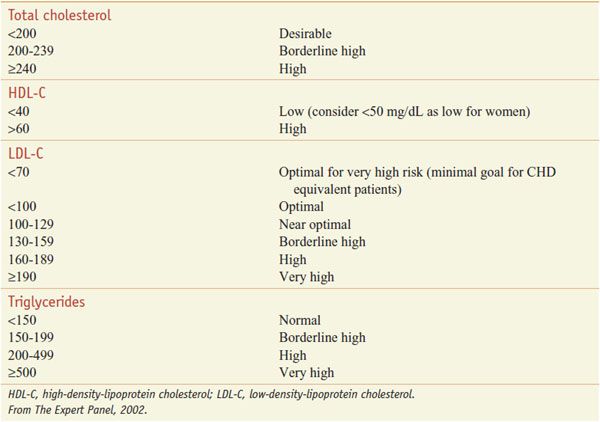
Table 31–5
Secondary Causes of Dyslipidemia
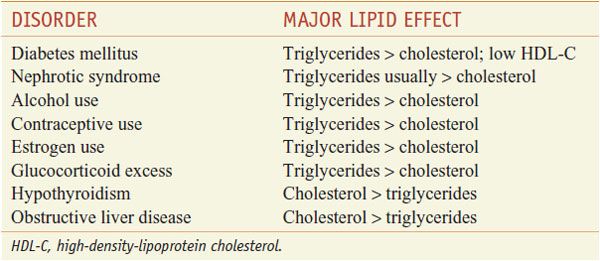
Table 31–6
Treatment Based on LDL-C Levels(2004 Revision of NCEP Adult Treatment Panel III Guidelines)
WHOM AND WHEN TO TREAT?
Large-scale trials with statins have provided new insights into which patients with dyslipidemia should be treated and when treatment should be initiated.
Gender. Both men and women benefit from lipid-lowering therapy. Statins are the recommended first-line drug therapy for lowering lipids and preventing CHD events in postmenopausal women.
Age. Age >45 years in men and >55 years in women is considered to be a CHD risk factor. The statin trials have shown that patients >65 years of age benefit from therapy as much as do younger patients. Old age per se is not a reason to refrain from initiating drug therapy in an otherwise healthy person.
Cerebrovascular Disease Patients. Plasma cholesterol levels correlate positively with the risk of ischemic stroke and statins reduce stroke and transient ischemic attacks in patients with and without CHD.
Peripheral Vascular Disease Patients. Statins are beneficial in patients with peripheral vascular disease.
Hypertensive Patients and Smokers. The risk reduction for coronary events in statin trials of hypertensive patients and smokers is similar to that in subjects without hypertension.
Type 2 Diabetes Mellitus. Patients with type 2 diabetes benefit very significantly from aggressive lipid lowering (see below).
Post–Myocardial Infarction or Revascularization Patients. As soon as CHD is diagnosed, it is essential to begin lipid-lowering therapy (NCEP guidelines: LDL-C goal <70 mg/dL for very high-risk patients). Statin therapy also improves the long-term outcome after bypass surgery.
TREATMENT OF TYPE 2 DIABETES PATIENTS
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


