Chapter 15
Drug discovery
15.1 Stages of drug discovery
- Target molecule
– selection
– validation
- Candidate compounds
– acquisition
– screening
Notes: Biochemical and physiological research suggests macromolecules that might make worthwhile targets for treating diseases. This is an important step toward new drugs, but only the first one. The subsequent steps that must be taken are the subject of this chapter.
Drug development is labor-intensive and costly, so it is important to experimentally ascertain whether a drug with the intended activity on our chosen target would indeed produce the expected physiological outcome. This stage of development is referred to as target validation.
The choice of the experimental model for target validation depends on the intended physiological effect. Some effects, for example cytotoxicity, can readily be observed in cell cultures; on the other hand, for physiological functions such as pain or blood pressure, animal experiments will be required. If the drug under consideration is supposed to be an inhibitor, its effect can often be modeled by genetic knockout of the target or by RNA interference (see slide 13.3.6). On the other hand, if the intention is to activate the target, it should in principle often be possible to model this by overexpression (cf. slide 2.6.1); however, this approach does not seem to be commonly used in practice.
If the target has been experimentally validated, candidate compounds must be obtained and screened. Major pharmaceutical companies have large libraries (collections) of compounds that may be screened over and over against novel, unrelated targets. Alternatively, custom libraries may be synthesized and can be structurally focused around some existing agonist or antagonist of the target molecule.
The screening of large numbers of compounds—a recent study on a specific type of voltage-gated K+ channels tested as many as 650,000 compounds [110]— requires simple and robust high-throughput assays. If detailed structural information on the macromolecular target is available, it may be preferable to perform the initial screening in silico, that is, to use molecular docking software to examine the binding of real or virtual compounds to the target.
Once lead compounds have been identified that act on the target in the intended manner, they must also be tested with other macromolecules that are related to the target; the entirety of such data for a given compound is its receptor profile. Lead compounds must also be tested against antitargets, that is, macromolecules that are frequently involved in drug toxicity. One important antitarget are cardiac KCNQ potassium channels, whose inhibition by drugs gives rise to cardiac arrhythmias.
15.1.1 Chemical structures of subtype-selective glutamate receptor ligands
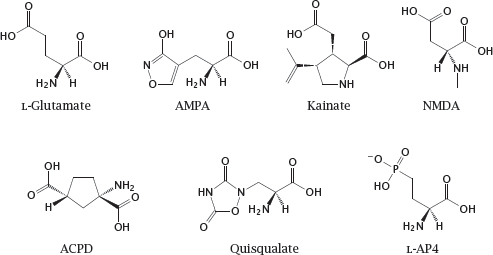
Acronyms: ACPD, 1-aminocyclopentane-1,3-dicarboxylic acid; AMPA, 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid; L-AP4, L-2-amino-4-phosphonobutanoic acid; NMDA, N-methyl-D-aspartate.
Notes: While a drug development project will usually start with a hypothesis that is based on the state of the art in biochemistry and physiology, one must nevertheless be ready to reevaluate this hypothesis as further experimental information accrues along the way. A common case in point is the discovery of pharmacologically distinct subtypes of receptors that at the outset were assumed to be homogeneous.
AMPA and quisqualate activate the same subtype of ionotropic glutamate receptors. Quisqualate also activates group I metabotropic glutamate receptors, which belong to the GPCR type. NMDA and kainate, respectively, activate two other ionotropic receptor subtypes. ACPD is an agonist of group I and II metabotropic glutamate receptors. L-AP4 is an agonist of group III metabotropic receptors (see section 6.11).
15.2 Sources of candidate compounds
- Synthetic libraries
- Natural compounds
- Semisynthesis
- Gene technology
Notes: Candidate compounds can be obtained from various sources. Natural compounds or synthetic strategies can be used alone or in combination; the latter approach is termed semisynthesis. Gene technology is a promising development but not yet a major source for compounds in practice.
15.2.1 Combinatorial synthesis: the Ugi reaction
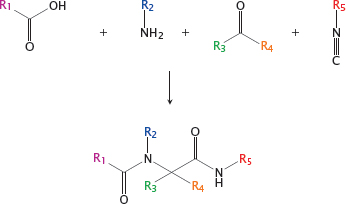
Notes: One important strategy for developing large compound libraries is referred to as combinatorial synthesis. In this approach, modular reactants that share similar reactive groups but differ in their side chains are combined in many permutations, all of which are processed in parallel, often with the help of robotic systems.
An example of a combinatorial synthetic strategy is the Ugi reaction. Here, a carboxylic acid, an amine, a ketone or aldehyde and an isonitrile condense to form a bisamide. Through combinatorial variation of the functional groups R1-R5, many different compounds can be prepared in parallel.
15.2.2 Semisynthesis of penicillins
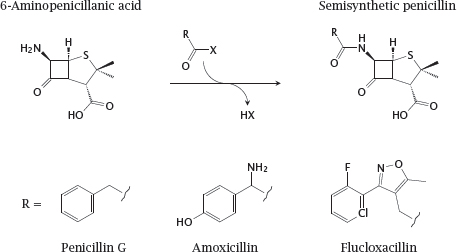
Notes: The penicillins are good examples for the semisynthetic variation of natural compounds. The starting compound, 6-aminopenicillanic acid, is obtained from fermenter cultures of the fungus Penicillium notatum, and is chemically acylated at its unique amino group.
15.2.3 Semisynthesis of cephalosporins
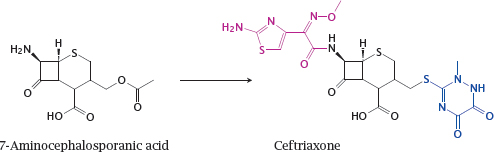
Notes: Ceftriaxone (shown here) and other cephalosporins (cf. slide 11.4.9) are derived through semisynthesis from 7-aminocephalosporanic acid. Here, two variable substituents are attached to the natural compound nucleus. More complex, multi-step chemistries are regularly utilized on a wide variety of natural products.
15.2.4 Biosynthesis of polyketides
Notes: Polyketides are natural compounds that are produced, in considerable variety, by Streptomyces species and other microorganisms. The biosynthesis of polyketides is chemically similar to that of fatty acids, with the following differences:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


