Disorders of Endocrine Function
Jacquelyn L. Banasik
Key Questions
• How can primary and secondary endocrine disorders be differentiated?
• What etiologic factors would lead to clinical manifestations of hormone excess or deficiency?

http://evolve.elsevier.com/Copstead/
Together with the nervous system, the endocrine system (the glands and the hormones they secrete) regulates body processes involving growth, maturation, metabolic functions, fluid balance, responses to stress, and reproduction.1,2 This regulation is carried out through the actions of the hormones produced and secreted by the endocrine cells. Endocrine hormones are chemical messengers that travel through the bloodstream to exert physiologic effects on specific target cells and tissues. In the healthy state, hormones are released by endocrine glands when their action is needed and inhibited when their effect is attained.
Endocrine disease is marked by either hyperfunction (excessively high blood concentrations of a hormone, or conditions that mimic high hormone levels) or hypofunction (depressed levels, or conditions that mimic low hormone levels). Some endocrine disorders have such striking characteristics that recognition is obvious. Other symptoms of endocrine disease may be nonspecific and more difficult to detect. Observing and interviewing skills are important because, with the exception of the thyroid and testicles, the endocrine glands cannot be directly examined. Laboratory diagnostic tests are especially important in assessing the endocrine system.
This chapter describes alterations in the anterior pituitary regulatory system, including growth hormone, thyroid hormone, and adrenal hormones, as well as parathyroid hormone disorders and posterior pituitary disorders of antidiuretic hormone (vasopressin) secretion. Disorders of prolactin and the gonadotropins (follicle-stimulating hormone and luteinizing hormone) are discussed in Unit IX. Disorders of insulin secretion are discussed in Chapter 41.
Basic Concepts of Endocrine Disorders
Etiology of Endocrine Disorders
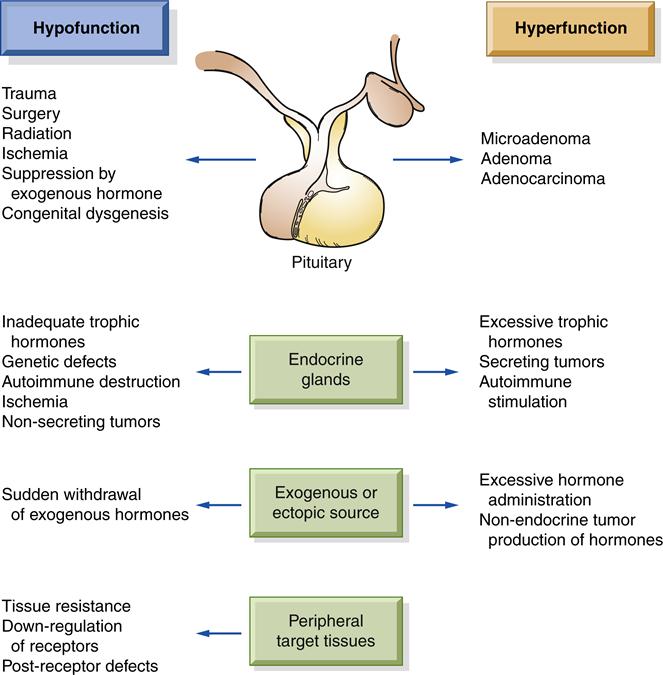
Dysfunction, either hyposecretion or hypersecretion, may originate in the hypothalamus/pituitary, the hormone-producing gland, or the target tissue (Figure 40-1). The etiology of endocrine disorders may be congenital, infectious, autoimmune, neoplastic, idiopathic, or iatrogenic. The onset of the disorders can be slow and insidious or abrupt and life threatening. The age at onset may range from birth to old age.
Abnormal hormone production occasionally results from an inborn genetic defect. Such defects may cause excessive production of hormone precursors because of an enzymatic block in the synthetic pathway, and enzyme deficiencies that impair hormone synthesis. An important example is congenital adrenal hyperplasia in which infants develop enlarged adrenal glands, but have a deficiency of cortisol production. If genetic defects do not cause a complete block of synthesis, increased pituitary stimulus may compensate by causing glandular hyperplasia, resulting in near-normal hormone levels.
Autoimmune disorders commonly cause endocrine dysfunction, particularly in women. The pathogenesis of autoimmunity is incompletely understood, but involves both a genetic predisposition and an environmental trigger (see Chapter 10). Antibodies are produced against certain antigens on self tissue cells, resulting either in hyperfunction of the endocrine gland (e.g., hyperthyroidism of Graves disease) or in immune destruction of the gland, eventually leading to hypofunction (e.g., adrenal insufficiency of Addison disease).
Hormones may be produced by abnormal tissue sites. Such ectopic hormone production is usually associated with a malignant tumor. Although different tumors can produce hormones, some cell types are more commonly associated with specific tumors. For example, some lung tumors produce antidiuretic hormone (ADH), leading to water intoxication and hyponatremia. Endocrine disorders can also be classified as functional disorders caused by nonendocrine disease such as chronic renal failure, liver disease, or heart failure.
In some cases endocrine disease occurs when the target tissue fails to respond to a hormone. The presence of normal or elevated hormone levels without normal hormonal action indicates target tissue resistance. This problem is also demonstrated by diminished or absent response to the administration of exogenous hormones. The mechanisms of hormone resistance may be genetic or acquired and may include defects at receptor sites, antibody reaction to hormone receptors, and defective postreceptor hormone action.
Finally, some endocrine disorders may be induced by medical treatments, such as therapy for a nonendocrine disorder. These iatrogenic disorders can be caused by chemotherapy, radiation therapy, or surgical removal of glands. Commonly, a treatment for endocrine hyperfunction involves removal or destruction of glandular tissue with resultant chronic hypofunction. Long-term hormone replacement therapy may then be needed.
Classification of Endocrine Disorders
Endocrine disorders involving control by the anterior pituitary gland commonly are classified as primary (intrinsic malfunction of the hormone-producing target gland) or secondary (malfunction of the hypothalamus/pituitary cells that control the hormone-producing target gland). The clinical presentation of an endocrine disorder of primary or secondary etiology is similar; however, diagnosing the source of the problem may be important in determining the best treatment. Measurement of serum concentrations of pituitary and target gland hormones allows differentiation between primary and secondary endocrine etiologies.
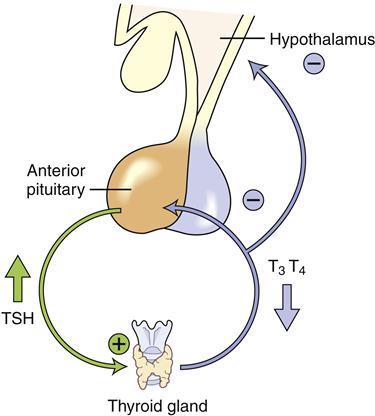
Clinically useful laboratory measures are available for diagnosing and monitoring hormone disorders including those affecting thyroid and adrenal function. Laboratory diagnosis is based on an understanding of the feedback loop communication between the hypothalamic-pituitary system and the target gland. When the primary gland fails, inadequate hormone is produced and low levels of hormone are present in the circulation, but blood levels of the corresponding trophic pituitary hormone become very elevated (Figure 40-2). For example, in primary hypothyroidism, the thyroid fails to secrete thyroid hormones and serum levels of thyroxine (T4) become lower. Thyroid-stimulating hormone (TSH) levels rise as the pituitary gland attempts to stimulate the malfunctioning thyroid. In contrast, in secondary hypothyroidism, the pituitary gland fails to release TSH, secondarily reducing thyroid gland production, so both thyroxine and TSH levels are abnormally low in the circulation. It is important to recall that the hormones released by the target gland are the ones that produce clinical signs and symptoms and that they are the starting point for interpretation of laboratory test results.
Growth Hormone Disorders
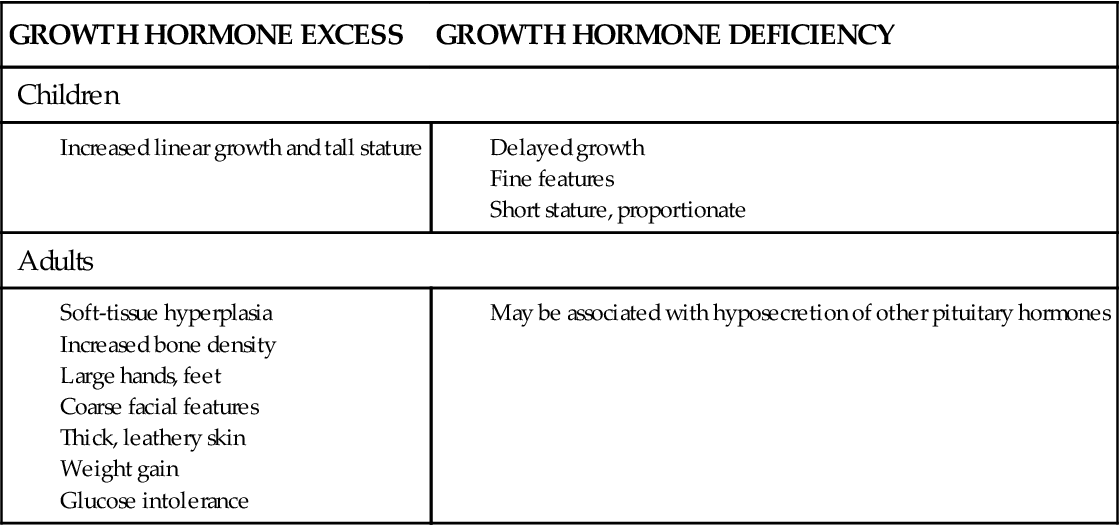
Growth hormone (GH) is produced in the anterior pituitary gland under the influence of hypothalamic releasing (growth hormone–releasing hormone) and inhibiting (somatostatin) factors. Its primary target organ is the liver, but GH also has direct effects on several tissue types. In general, GH increases lean body mass, reduces fat mass, and induces the liver to release glucose under conditions of hypoglycemia. Many of the effects of GH are mediated by a peptide called IGF-1 (insulin-like growth factor-1) that is released from the liver when stimulated by GH. Please refer to Chapter 39 for details of GH synthesis, regulation, and activity. The major signs and symptoms of GH imbalance are summarized in Box 40-1.
BOX 40-1
SIGNS AND SYMPTOMS OF GROWTH HORMONE IMBALANCE
Growth Hormone Deficiency
Etiology and pathogenesis
Deficiencies in GH secretion can be classified into several major categories: (1) decreased GH secretion, (2) defective GH action (structurally abnormal GH or defective GH receptor), and (3) defective IGF-1 (somatomedin) generation.
Growth hormone deficiency is most clinically relevant in children. A birth history of prolonged labor or breech delivery is common, but GH deficiency may also be present in children who are born with midline craniocerebral defects, most likely attributable to congenital malformations or as sequelae of a chromosomal anomaly. Deficiencies in GH and other pituitary hormones should be considered in any child with nystagmus, retinal abnormalities, and other midline or midfacial abnormalities, such as cleft lip or palate. The association between GH deficiency and other midline abnormalities appears to occur because the pituitary gland is developing during the same stage of fetal life as the other midline structures.
Children with GH deficiency have a variety of presentations, depending on the cause of the deficiency, the age at onset, and the severity of the disorder. The basis for this defect may be failure of the hypothalamus to stimulate pituitary GH secretion or failure of the pituitary to produce GH.
The most common tumors to influence hypothalamic-pituitary function are midline brain tumors. These include gliomas of the optic nerve and craniopharyngiomas. Craniopharyngiomas arise from cells at the junction of the anterior and posterior pituitary gland, are believed to be present at birth, and are slow growing. Craniopharyngiomas may grow to a large size without producing typical signs of increased intracranial pressure (vomiting, headache, oculomotor abnormalities). In older children, delayed growth may be the first symptom of a craniopharyngioma. Radiation therapy for brain tumors or leukemia may cause damage to hypothalamic and pituitary function. Traumatic insult to the skull or sella turcica may damage the pituitary gland, interrupting vascular connections and hypothalamic stimulation.
Clinical manifestations
GH-deficient infants usually have normal birth length and weight. They may manifest hypoglycemia because GH and cortisol are necessary to maintain the euglycemic state. Hypoglycemia may present after fasting, which could be as brief as 3 hours in an infant. Recurrent episodes of hypoglycemia may lead to seizures and permanent cerebral damage. Boys developing GH deficiency in utero may have micropenis and undescended testicles.
GH-deficient children fall below the third percentile of growth in comparison with their peers. Dental eruption is delayed, and the development and setting of the permanent teeth are irregular. The hair is thin, and the nail growth is poor. Older children have greater fat mass and decreased muscle mass, with delayed bone formation. Delayed puberty is common if other anterior pituitary hormones are also affected.
Children’s growth should be evaluated annually. If growth velocity is abnormal, an endocrinologic evaluation and physiologic tests to stimulate GH release can be planned. Many pharmacologic agents are available that stimulate GH secretion in children, including insulin, arginine, levodopa, and clonidine. Children’s neurosecretory GH patterns can be studied by obtaining timed serum GH samples during a normal nighttime sleep cycle.
Treatment
Hormonal replacement therapy for GH-deficient children has been available for several decades. The most obvious effect of GH replacement is stimulation of linear growth in children whose bones have not fused. With treatment, children experience an increase in growth velocity as well as depletion of the excess fat stores noted in GH-deficient children.
Acquired GH deficiency in adults has only recently been recognized and treatment approved by the Food and Drug Administration.3 Adults may become GH deficient after resection of pituitary tumors or after traumatic head injuries. There is controversy regarding the manifestations of GH deficiency acquired in adulthood. There appears to be increased mortality attributable to cardiovascular causes when adults do not receive GH replacement following pituitary damage. GH-deficient adults may have diminished lean body mass, hypercholesterolemia, and decreased bone density.
Growth Hormone Excess
Etiology and pathogenesis
GH excess is nearly always due to uncontrolled production of the hormone by a benign somatotropic tumor in the pituitary gland adenoma. GH stimulates the liver to produce IGF-1, and these two hormones act in concert to cause up-regulated growth of soft and bony tissues. Because GH secretion varies significantly over the course of the day, the serum level of IGF-1, which is more stable, may be measured as an indicator of GH secretion. An elevated IGF-1 level is a useful indicator of GH hypersecretion. If the tumor presents in childhood before the skeletal epiphyses are closed, rapid growth results in pituitary giantism. These children experience markedly accelerated growth velocity and quickly exceed the 95th percentile on pediatric growth charts. When the disorder is allowed to progress untreated, some of these children may grow to 8 feet or more in height and usually suffer an early cardiovascular death related to cardiomegaly and heart failure.
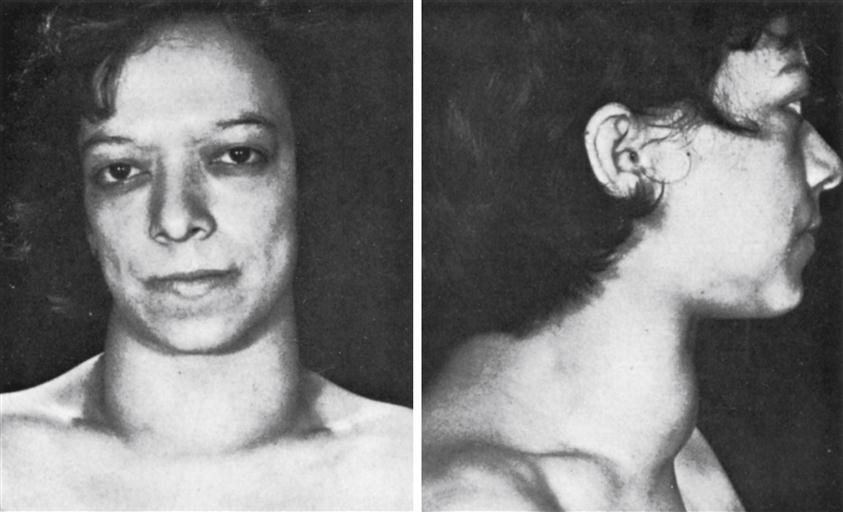
In adults, GH excess is called acromegaly and may be clinically subtle. Acromegaly occurs with equal frequency in men and women during the fourth and fifth decades of life. After the skeletal epiphyses close, bony growth increases bone density and thickening of the short bones, such as the hands and feet.
Clinical manifestations of acromegaly
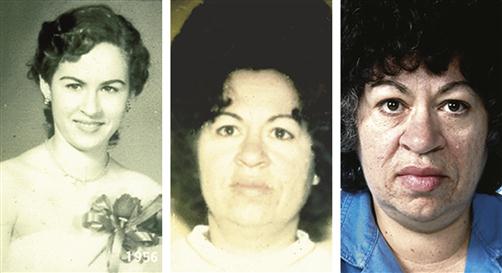
Patients usually notice increased ring and shoe size, which progressively advances over several years. Enlargement of the frontal sinus causes a prominent brow, and growth of the mandible results in progressive underbite (prognathism) (Figure 40-3). Soft tissues also slowly hypertrophy, causing coarsening of facial features and skin tags. Internal organs increase in size, resulting in goiter (thyroid enlargement) and cardiomegaly. Other manifestations include deepening of the voice secondary to vocal cord thickening and enlargement of the tongue, resulting in sleep apnea. Colonic polyps become more common, with the potential for malignant degeneration. It is estimated that the average patient with acromegaly has an active pituitary tumor for 7 or more years before seeking evaluation. Most often the changes are attributed by the patient and his or her family as “just growing older.” Abnormalities in bone and soft-tissue growth are mostly irreversible. Patients may develop symptoms of increased intracranial pressure if the pituitary tumor enlarges significantly, including headache and visual disturbances. Some patients may develop glucose intolerance or hyperglycemia, and GH has been called a diabetogenic hormone for this reason.
Treatment
Effective therapy for acromegaly involves surgically removing the tumor while counteracting the effects of excess GH. Octreotide, a synthetic form of somatostatin, suppresses production of GH. Surgery is usually performed using a transsphenoidal approach, but often the tumor is too large to completely resect. Radiation may be used postoperatively in these cases.
Thyroid Hormone Disorders
Secretion of the thyroid hormones, triiodothyronine (T3) and thyroxine (T4), is under control of thyroid-stimulating hormone (TSH) secretion from the anterior pituitary gland. In turn, TSH secretion from the pituitary is under control of thyroid-releasing hormone (TRH) from the hypothalamus. Thyroid hormones are important for normal growth and development of tissues throughout the body and an important regulator of metabolism. The details of thyroid hormone synthesis, regulation, and activity are described in Chapter 39.
Hypothyroidism
Etiology and pathogenesis
Hypothyroidism may be congenital in origin or acquired later in life. The great majority of cases of hypothyroidism are primary, due to intrinsic dysfunction of the thyroid gland. Congenital hypothyroidism may result from a variety of causes. Thyroid dysgenesis (lack of thyroid gland development) accounts for most of the cases of congenital hypothyroidism. Abnormal TSH receptors and defective synthesis of thyroid hormone are other mechanisms causing congenital hypothyroidism.4 Congenital hypothyroidism that results in significant defects in mental and physical development may be referred to as cretinism.
Lymphocytic thyroiditis (Hashimoto thyroiditis, or autoimmune thyroiditis) is the most common cause of acquired hypothyroidism. Lymphocytic thyroiditis is characterized by an enlarged thyroid gland (Figure 40-4) caused by lymphocytic infiltration. Thyroid hormone production decreases, stimulating the release of TSH from the pituitary gland and resulting in elevated serum TSH levels. Hypothyroidism and its clinical symptoms progress as the gland becomes fibrotic.
Other causes of acquired hypothyroidism include irradiation of the thyroid gland, surgical removal of thyroid tissue, and iodine deficiency. Iodine is essential for the formation of T4 and T3. Lack of iodine prevents production of both T4 and T3 but does not stop the formation of thyroglobulin. As a result, insufficient hormone is available to inhibit production of TSH by the anterior pituitary gland. The elevated TSH level then causes the thyroid cells to secrete excessive amounts of thyroglobulin (colloid) into the follicles, and the gland grows larger and larger, producing a goiter (see Figure 40-4). An enlarged thyroid (goiter) is not always associated with hypothyroidism and can be present in euthyroid and hyperthyroid states.
Some foods contain “goitrogenic” substances that interfere with thyroid hormone synthesis. Such goitrogenic substances occur in some varieties of turnips and cabbage, but the clinical significance of these goitrogens is considered to be minimal. The drug lithium inhibits thyroid hormone synthesis and secretion and causes hypothyroidism in up to 20% of patients.5
Secondary hypothyroidism is caused by defects in TSH production and is uncommon. Individuals who have been exposed to severe head trauma, cranial neoplasms, brain infections, cranial irradiation, and neurosurgery can be left with secondary hypothyroidism.
Clinical manifestations
Routine screening of newborns has resulted in early treatment of most infants with congenital hypothyroidism. Few clinical manifestations are present at birth. In untreated infants, symptoms appear in the first months of life and include a dull appearance; a thick, protuberant tongue; and thick lips (leading to feeding difficulties). Other signs include prolonged neonatal jaundice, poor muscle tone, bradycardia, mottled extremities, umbilical hernia, and a hoarse cry. Thyroid hormone is essential for normal central nervous system development; significant and irreversible intellectual disability will occur unless thyroid hormone replacement therapy is started early in infancy. Older children who acquire hypothyroidism have essentially the same clinical manifestations as seen in adults. In addition, growth retardation, delayed bone development, and delayed or precocious puberty may occur.
In general, individuals with hypothyroidism have decreased basal metabolic rates as the basis for many of their signs and symptoms. Patients report subjective feelings of weakness, lethargy, cold intolerance, and decreased appetite. Bradycardia, narrowed pulse pressure, and mild to moderate weight gain may occur. Elevated levels of serum cholesterol and triglycerides are common as is an increased incidence of atherosclerosis. The thyroid gland may become enlarged (goitrous), the skin may be cool and dry, and constipation may be present. Depression and difficulties with concentration and memory occur. Women with acquired hypothyroidism may experience menstrual irregularities, with increased flow and clotting.6 Box 40-2 summarizes the general signs and symptoms of thyroid imbalance.
BOX 40-2
TYPICAL SIGNS AND SYMPTOMS OF THYROID IMBALANCE



