Chapter 20 Diabetes mellitus and other disorders of metabolism
Diabetes mellitus
Hyperglycaemia, insulin and insulin action
Insulin structure and secretion
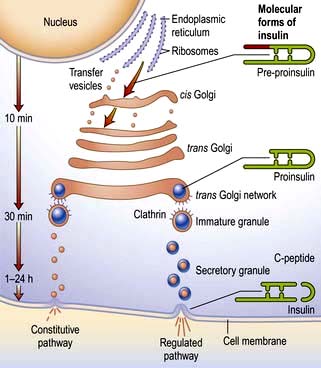
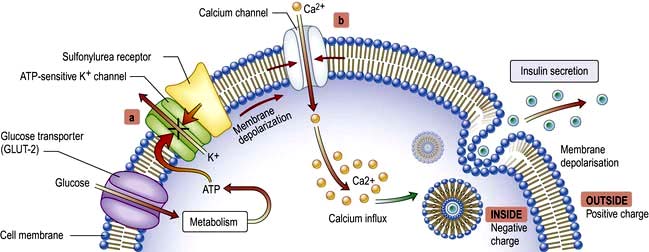
Insulin is the key hormone involved in the storage and controlled release within the body of the chemical energy available from food. It is coded for on chromosome 11 and synthesized in the beta cells of the pancreatic islets (Fig. 20.1). The synthesis, intracellular processing and secretion of insulin by the beta cell is typical of the way that the body produces and manipulates many peptide hormones. Figure 20.2 illustrates the cellular events triggering the release of insulin-containing granules. After secretion, insulin enters the portal circulation and is carried to the liver, its prime target organ. About 50% of secreted insulin is extracted and degraded in the liver; the residue is broken down by the kidneys. C-peptide is only partially extracted by the liver (and hence provides a useful index of the rate of insulin secretion) but is mainly degraded by the kidneys.
An outline of glucose metabolism
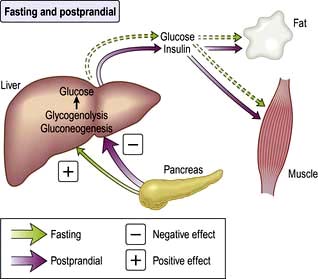
Insulin is a major regulator of intermediary metabolism, although its actions are modified in many respects by other hormones. Its actions in the fasting and postprandial states differ (Fig. 20.3). In the fasting state, its main action is to regulate glucose release by the liver, and in the postprandial state, it additionally promotes glucose uptake by fat and muscle. The effect of counter-regulatory hormones (glucagon, epinephrine (adrenaline), cortisol and growth hormone) is to cause greater production of glucose from the liver and less utilization of glucose in fat and muscle for a given level of insulin.
 GLUT-1 – enables basal non-insulin-stimulated glucose uptake into many cells (see Fig. 6.29).
GLUT-1 – enables basal non-insulin-stimulated glucose uptake into many cells (see Fig. 6.29).

 GLUT-3 – enables non-insulin-mediated glucose uptake into brain neurones and placenta.
GLUT-3 – enables non-insulin-mediated glucose uptake into brain neurones and placenta.
 GLUT-4 – enables much of the peripheral action of insulin. It is the channel through which glucose is taken up into muscle and adipose tissue cells following stimulation of the insulin receptor (Fig. 20.4).
GLUT-4 – enables much of the peripheral action of insulin. It is the channel through which glucose is taken up into muscle and adipose tissue cells following stimulation of the insulin receptor (Fig. 20.4).
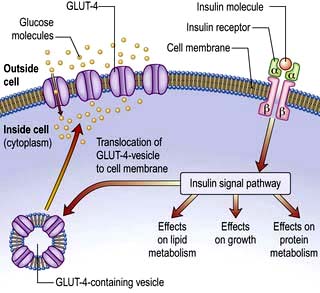
This is a glycoprotein (400 kDa), coded for on the short arm of chromosome 19, which straddles the cell membrane of many cells (Fig. 20.4). It consists of a dimer with two α-subunits, which include the binding sites for insulin, and two β-subunits, which traverse the cell membrane. When insulin binds to the α-subunits it induces a conformational change in the β-subunits, resulting in activation of tyrosine kinase and initiation of a cascade response involving a host of other intracellular substrates. One consequence of this is migration of the GLUT-4 glucose transporter to the cell surface and increased transport of glucose into the cell. The insulin-receptor complex is then internalized by the cell, insulin is degraded, and the receptor is recycled to the cell surface.
Classification of diabetes
Diabetes may be primary (idiopathic) or secondary (Table 20.1). Primary diabetes is classified into:


Table 20.1 Aetiological classification of diabetes mellitus, based on classification by the American Diabetes Association (ADA)
Note: Patients with any form of diabetes may require insulin treatment at some stage of their disease. Such use of insulin does not, of itself, classify the patient.
(Adapted from ADA. Diagnosis and classification of diabetes mellitus. Diabetes Care 2008; 31(Suppl 1):S55–S60.)
The key clinical features of the two main forms of diabetes are listed in Table 20.2. Type 1 and type 2 diabetes represent two distinct diseases from the epidemiological point of view, but clinical distinction can sometimes be difficult. The two diseases should from a clinical point of view be seen as a spectrum, distinct at the two ends but overlapping to some extent in the middle. Hybrid forms are increasingly recognized, and patients with immune-mediated diabetes (type 1) may, for example, also be overweight and insulin resistant. This is sometimes referred to as ‘double diabetes’. It is more relevant to give the patient the right treatment on clinical grounds than to worry about how to label their diabetes. The classification of primary diabetes continues to evolve. Monogenic forms have been identified (see p. 1007), in some cases with significant therapeutic implications. Although secondary diabetes accounts for barely 1–2% of all new cases at presentation, it should not be missed because the cause can sometimes be treated. All forms of diabetes derive from inadequate insulin secretion relative to the needs of the body, and progressive insulin secretory failure is characteristic of both common forms of diabetes. Thus, some patients with immune-mediated diabetes type 1 may not at first require insulin, whereas many with type 2 diabetes will eventually do so.
Table 20.2 The spectrum of diabetes: a comparison of type 1 and type 2 diabetes mellitus
| Type 1 | Type 2 | |
|---|---|---|
Age | Younger (usually <30) | Older (usually >30) |
Weight | Lean | Overweight |
Symptom duration | Weeks | Months/years |
Higher risk ethnicity | Northern European | Asian, African, Polynesian and American-Indian |
Seasonal onset | Yes | No |
Heredity | HLA-DR3 or DR4 in >90% | No HLA links |
Pathogenesis | Autoimmune disease | No immune disturbance |
Ketonuria | Yes | No |
Clinical | Insulin deficiency | Partial insulin deficiency initially |
| ± ketoacidosis | ± hyperosmolar state |
| Always need insulin | Need insulin when beta cells fail over time |
Biochemical | C-peptide disappears | C-peptide persists |
Type 1 diabetes mellitus
Epidemiology
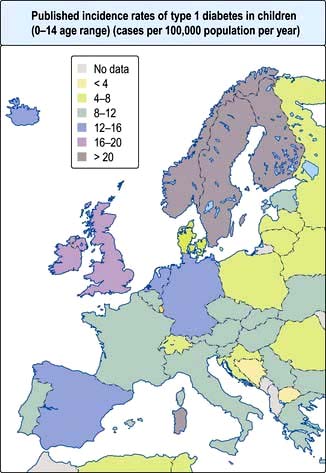
Type 1 diabetes is a disease of insulin deficiency. In western countries almost all patients have the immune-mediated form of the disease, otherwise known as type 1A. Type 1 diabetes is a disease of childhood, reaching a peak incidence around the time of puberty, but can present at any age. A ‘slow-burning’ variant with slower progression to insulin deficiency occurs in later life and is sometimes called latent autoimmune diabetes in adults (LADA). LADA may be difficult to distinguish from type 2 diabetes. Clinical clues are: leaner build, rapid progression to insulin therapy following an initial response to other therapies, and the presence of circulating islet autoantibodies. The highest rates of type 1 diabetes in the world are seen in Finland and other Northern European countries, and on the island of Sardinia, which for unknown reasons, has the second highest rate in the world (Fig. 20.5). The incidence of type 1 diabetes appears to be increasing in most populations. In Europe, the annual increase is of the order of 2–3%, and is most marked in children under the age of 5 years. WHO estimated in 1995 that there were 19.4 million people with type 1 diabetes and that the number will rise to 57.2 million by 2025.
Causes
Autoimmunity and type 1 diabetes
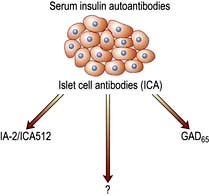
Type 1 diabetes is associated with other organ-specific autoimmune diseases including autoimmune thyroid disease, coeliac disease, Addison’s disease and pernicious anaemia. Autopsies of patients who died following diagnosis of type 1 diabetes show infiltration of the pancreatic islets by mononuclear cells. This appearance, known as insulitis, resembles that in other autoimmune diseases such as thyroiditis. Several islet antigens have been characterized, and these include insulin itself, the enzyme glutamic acid decarboxylase (GAD), protein tyrosine phosphatase (IA-2) (Fig. 20.6) and the cation transporter ZnT8. Recent studies have shown that GAD immunotherapy has no benefit. The observation that treatment with immunosuppressive agents such as ciclosporin prolongs beta-cell survival in newly diagnosed patients has confirmed that the disease is immune-mediated.
Environmental factors
The incidence of childhood type 1 diabetes is rising across Europe at the rate of 2–3% each year, suggesting that environmental factor(s) are involved in its pathogenesis. Islet autoantibodies (see above) appear in the first few years of life, indicating prenatal or early postnatal interactions with the environment. Exposures to dietary constituents, enteroviruses such as Coxsackie B4 and relative deficiency of vitamin D are possible candidates, but their role in the causation of the disease has yet to be confirmed. A cleaner environment with less early stimulation of the immune system in childhood may increase susceptibility for type 1 diabetes, as for atopic/allergic conditions (the hygiene hypothesis) (see p. 824), and more rapid weight gain in childhood and adolescence leading to increased insulin resistance might accelerate clinical onset (the accelerator hypothesis).
Type 2 diabetes mellitus
Epidemiology
Type 2 diabetes is associated with central obesity, hypertension, hypertriglyceridaemia, a decreased HDL-cholesterol, disturbed haemostatic variables and modest increases in a number of pro-inflammatory markers. Insulin resistance is strongly associated with many of these variables, as is increased cardiovascular risk. This group of conditions is referred to as the metabolic syndrome (see p. 223). The International Diabetes Federation has proposed criteria based on increased waist circumference (or BMI >30) plus two of the following: diabetes (or fasting glucose >6.0 mmol/L), hypertension, raised triglycerides or low HDL cholesterol. On this definition, about one-third of the adult population has features of the syndrome, not necessarily associated with diabetes. Critics would argue that the metabolic syndrome is not a distinct entity, but one end of a continuum in the relationship between exercise, lifestyle and bodyweight on the one hand, and genetic make-up on the other, and that diagnosis adds little to standard clinical practice in terms of diagnosis, prognosis or therapy.
Causes
Abnormalities of insulin secretion and action
The relative role of secretory failure versus insulin resistance in the pathogenesis of type 2 diabetes has been much debated, but even massively obese individuals with a fully functioning beta-cell mass do not necessarily develop diabetes, which implies that some degree of beta-cell dysfunction is necessary. Insulin binds normally to its receptor on the surface of cells in type 2 diabetes, and the mechanisms of ‘insulin resistance’ are still poorly understood. Insulin resistance is, however, associated with central obesity and accumulation of intracellular triglyceride in muscle and liver in type 2 diabetes, and a high proportion of patients have non-alcoholic fatty liver disease (NAFLD), see page 303. It has long been stated that patients with type 2 diabetes retain up to 50% of their beta-cell mass at the time of diagnosis, as compared with healthy controls, but the shortfall is greater than this when they are matched with healthy individuals who are equally obese. In addition, patients with type 2 diabetes almost all show islet amyloid deposition at autopsy, derived from a peptide known as amylin or islet amyloid polypeptide (IAPP), which is co-secreted with insulin. It is not known if this is a cause or consequence of beta-cell secretory failure.
Monogenic diabetes mellitus
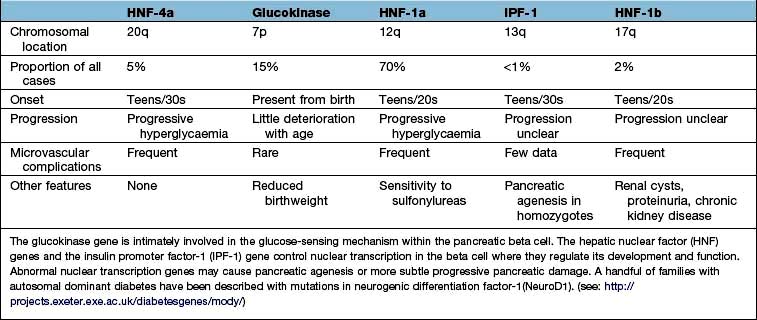
The genetic causes of some rare forms of diabetes are shown in Table 20.3. Considerable progress has been made in understanding these rare variants of diabetes. Genetic defects of beta-cell function (previously called ‘maturity-onset diabetes of the young’, MODY) are dominantly inherited, and several variants have been described, each associated with different clinical phenotypes (Table 20.4). These should be considered in people presenting with early-onset diabetes in association with an affected parent and early-onset diabetes in ~50% of relatives. They can often be treated with a sulfonylurea.
Table 20.3 Rare genetic causes of type 2 diabetes
| Disorder | Features |
|---|---|
Insulin receptor mutations | Obesity, marked insulin resistance, hyperandrogenism in women, acanthosis nigricans (areas of hyperpigmented skin) |
Maternally inherited diabetes and deafness (MIDD) | Mutation in mitochondrial DNA. Diabetes onset before age 40. Variable deafness, neuromuscular and cardiac problems, pigmented retinopathy |
Wolfram’s syndrome (DIDMOAD – diabetes insipidus, diabetes mellitus, optic atrophy and deafness) | Recessively inherited. Mutation in the transmembrane gene, WFS1. Insulin-requiring diabetes and optic atrophy in the first decade. Diabetes insipidus and sensorineural deafness in the second decade progressing to multiple neurological problems. Few live beyond middle age |
Severe obesity and diabetes | Alström’s, Bardet–Biedl and Prader–Willi syndromes. Retinitis pigmentosa, mental insufficiency and neurological disorders |
Disorders of intracellular insulin signalling. All with severe insulin resistance | Leprechaunism, Rabson–Mendenhall syndrome, pseudoacromegaly, partial lipodystrophy: lamin A/C gene mutation |
Genetic defects of beta-cell function | See Table 20.4 |
FURTHER READING
Chan JC, Malik V, Jia W et al. Diabetes in Asia: epidemiology, risk factors and pathophysiology. JAMA 2009; 301:2129–2140.
Ramachandran A, Ma RC, Snehalatha C. Diabetes in Asia. Lancet 2010; 375:408–418.
Sturnvoll M, Goldstein BJ, van Haefken TW. Type 2 diabetes; pathogenesis and treatment. Lancet 2008; 371:2153–2156.
Clinical presentation of diabetes
Presentation may be acute, subacute or asymptomatic.
Acute presentation
Young people often present with a 2–6-week history and report the classic triad of symptoms:
 Polyuria – due to the osmotic diuresis that results when blood glucose levels exceed the renal threshold
Polyuria – due to the osmotic diuresis that results when blood glucose levels exceed the renal threshold
 Thirst – due to the resulting loss of fluid and electrolytes
Thirst – due to the resulting loss of fluid and electrolytes






Asymptomatic diabetes
Physical examination at diagnosis
Evidence of weight loss and dehydration may be present, and the breath may smell of ketones. Older patients may present with established complications, and the presence of the characteristic retinopathy is diagnostic of diabetes. In occasional patients, there will be physical signs of an illness causing secondary diabetes (see Table 20.1). Patients with severe insulin resistance may have acanthosis nigricans, which is characterized by blackish pigmentation at the nape of the neck and in the axillae (p. 1217).
Diagnosis and investigation of diabetes
Diabetes is easy to diagnose when overt symptoms are present, and a glucose tolerance test is hardly ever necessary for clinical purposes. The oral glucose tolerance test has, however, allowed more detailed epidemiological characterization based on the existence of separate glucose thresholds for macrovascular and microvascular disease. These correspond with the levels for the diagnosis of impaired glucose tolerance (IGT) and diabetes as specified by the WHO criteria set out in Box 20.1. Epidemiological studies show that for every person with known diabetes, there is another undiagnosed in the population. A much larger proportion fall into the intermediate category of impaired glucose tolerance.
 Box 20.1
Box 20.1






