Relatively small kidneys of small for gestational age and prematurely born infants may have nephron deficit, a condition known as congenital oligonephropathy, which could lead to renal functional problems in adulthood. Long-term epidemiologic studies of these small-for gestational-age infants demonstrate an increased incidence of cardiovascular disease, hypertension, hyperlipidemia, diabetes mellitus, and renal failure in adulthood (27).
Another form of developmental hypoplasia is the unirenicular or unipapillary kidney, an uncommon developmental abnormality that is usually unilateral (28). Histopathologic studies show normally developed parenchyma, with focal glomerular sclerosis. Bilateral involvement results in the same clinical characteristics and the same evolution as oligonephronic hypoplasia.
Oephrolithiasis. The frequency of hypertension is not known. Prenatal and neonatal ultrasonography may be diagnostic for renal hypoplasia (29).
SIMPLE RENAL HYPOPLASIA
Simple renal hypoplasia, which consists of a smaller number of structurally normal nephrons, is a distinct entity. This may be bilateral or, less commonly, unilateral. It differs from oligonephronic hypoplasia in that nephronic hypertrophy is not a feature and a reduction in renal mass is generally less extreme.
RENAL SEGMENTAL ATROPHY (SEGMENTAL HYPOPLASIA, ASK-UPMARK KIDNEY)
Despite continuing controversy, the principal cause of small kidneys with segmental or lobar atrophy is vesicoureteric reflux with intrarenal reflux (30–33). Segmental hypoplasia is an outmoded concept, and the term Ask-Upmark kidney can be dropped from the lexicon. However, not all patients with this abnormality have demonstrable vesicoureteric reflux at the time of clinical evaluation, and other causes have been considered, such as localized vascular insufficiency. The possibility of localized developmental arrest was suggested in early descriptions of the abnormality (34), and that explanation still receives support. One of the arguments advanced in its favor is the occurrence of renal dysplasia in the abnormal segments (35), but the altered metanephric development is probably a consequence of intrauterine reflux. Serial radiographic studies have demonstrated that the segmental lesion initially develops in unscarred kidneys of normal size (36). The frequent paucity of inflammatory cells in the abnormal segments, as well as in the remainder of the kidney, has been taken as evidence that the lesion could not have arisen from reflux and chronic pyelonephritis. However, inflammatory infiltrates undoubtedly clear, so its causal effects could go unrecognized. This condition also needs to be evaluated in relation to sterile intrarenal reflux and its consequences.
The characteristic abnormality is a shrunken lobe containing an attenuated cortex and an effaced medullary pyramid. The cortex typically seems to lack glomeruli and to consist of microcystic tubules plugged with colloid casts, so-called aglomerular hypoplasia (Fig. 40.2). A few specimens contain easily recognizable collapsed glomeruli, indicating that the obsolete glomeruli and eventually the tubules are resorbed to leave a fibrous scar without identifiable nephrons (32). The arcuate and interlobular arteries are, until the end-stage fibrous scar, prominent as the result of cortical shrinkage and their own medial hypertrophy. Cavernous, thin-walled vessels can be traced into the renal sinus, where, despite their resemblance to dilated lymphatics, they connect with renal vein tributaries. The medullary pyramids contain a reduced number of ducts and a paucity or absence of vasa recta and recurrent loops. The ducts sometimes have a primitive appearance, taken as evidence of renal dysplasia (Fig. 40.2). These features are typically found in very small kidneys, partly accounting for the interpretation that the kidneys are hypoplastic; the kidneys have weights of 40 g or less, even in adult patients.
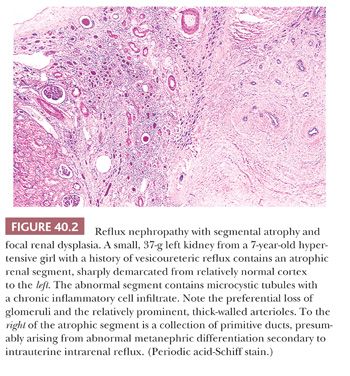
The presence of lobar or segmental atrophy in pyelonephritic and in dysplastic kidneys can be proof of reflux nephropathy. The abnormal segments in kidneys damaged by reflux are initially at the poles, with later involvement of middle segments. The remnant kidney undergoes compensatory hypertrophy, sometimes with a deceptively bland histopathologic appearance, overshadowing the adjacent shrunken lobes. Segmental glomerular sclerosis in hypertrophied glomeruli of the remnant kidney is associated with proteinuria, sometimes in the nephrotic range, and glomerular sclerosis is an important factor in progression to end-stage renal failure (37–39). The other important factor is frequent bilaterality of the reflux.
Many of the patients come to medical attention because of hypertension, not always with a history of recurrent urinary tract infection. The evaluation of nephrectomy specimens from patients with renal hypertension may seem to show only a small kidney with normal parenchyma, and thorough examination of the entire pyelocalyceal system is necessary to identify shrunken and atrophic segments.
CONGENITAL NEPHROMEGALY
Compensatory growth and hypertrophy of one kidney can occur when the contralateral kidney is severely diseased, dysplastic, or congenitally absent. Renal enlargement from an increased amount of renal parenchyma occurs in association with the Wiedemann-Beckwith syndrome (exomphalos, macroglossia, and gigantism) (40) and Perlman syndrome (macrosomia, islet-cell hypertrophy, unusual facies, and renal hamartomas) (41,42). The kidneys in both syndromes are excessively lobulated and contain dysplastic medullary pyramids, sometimes with small cysts. The medullary abnormality has often been characterized clinically and radiographically as medullary sponge kidney (43), despite the clear morphologic evidence of dysplasia and the clear differences from typical medullary sponge kidney. Histopathologic examination of cortical tissue reveals persistent nephrogenesis, nodular blastema, and nephroblastomatosis. The occurrence of nephroblastomatosis imposes an increased risk of Wilms tumor, as seen also in hemihypertrophy syndromes. Wiedemann-Beckwith syndrome carries an increased risk of adrenal carcinoma, hepatoblastoma, and brainstem glioma (44). Wiedemann-Beckwith syndrome is a dominant mutation, whereas Perlman syndrome is autosomal recessive. Nonetheless, some of the Wiedemann-Beckwith manifestations, for example, umbilical hernia and macroglossia, have been seen to develop postnatally (45).
SUPERNUMERARY KIDNEY
Rarely, a third kidney is present. It generally has an independent pelvicalyceal system that drains separately into the ureter; in some cases, complete duplication of the ureter occurs. The extra kidney is usually much smaller than the other two and is situated in the renal fossa below a normal kidney. Very rarely, it could be situated in a remote location such as the scrotum (46) or near the common bile duct (47).
RENAL DYSPLASIA
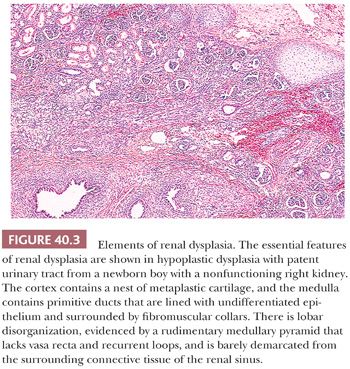
Dysplastic kidneys are, by definition, abnormally differentiated, as shown by abnormal structural organization with abnormally developed metanephric elements. The features that can be regarded as clearly dysplastic are metaplastic cartilage, primitive ducts, and lobar disorganization (Fig. 40.3) (17,48). Metaplastic cartilage customarily appears within the cortex as bars and nests of hyaline cartilage. Primitive ducts, which may be cystic, are altered collecting ducts lined with undifferentiated epithelium and surrounded by fibromuscular collars. Incomplete and abnormal corticomedullary relationships and rudimentary medullary development constitute lobar disorganization. The incompletely developed medullary pyramids are deficient in vasa recta and Henle loops and are associated with incomplete calyceal and forniceal development. These renal abnormalities bear a strong relationship to other urinary tract malformations, including ureteral atresia and urethral valves, suggesting that urinary obstruction or urinary reflux during metanephric development leads to renal dysplasia (17). In the early stages of multicystic dysplastic kidney, normal nephrogenesis occurs in what seems to be a normal metanephric blastema; however, an intrinsic abnormality in the branching morphogenesis of the ureteric duct might be responsible for the development of the histopathologic changes (49). This association with urinary tract abnormalities is seen in approximately 90% of cases; in the remaining 10%, the occurrence of hereditary and syndromal dysplasia, unaccounted for by urinary tract obstruction, implicates other pathogenetic factors.
Dysplastic kidneys are often cystic, and the most common variety perhaps is the multicystic kidney (50). Multicystic kidney dysplasia (MCKD), characterized by an enlarged, misshapen, irregularly cystic kidney (Fig. 40.4), is closely related to aplastic dysplasia, characterized by a small, barely recognizable, rudimentary, solid nubbin. The difference is in the degree of cyst formation, and all degrees intermediate between the two prototypes exist. The seemingly disorganized structure of the multicystic kidney is accounted for by the severity of cyst formation, and both multicystic and aplastic kidneys contain rudimentary lobes and lobules of metanephric tissue, with variable deficiencies of nephrons and ducts. Both contain relatively solid central areas, from which branching primitive ducts radiate to the periphery as incompletely differentiated branches of the ureteric bud. The septa among the cysts in multicystic kidneys contain rudimentary lobules consisting of branching collecting ducts in close relation to caps of cortical glomeruli and convoluted tubules. Cysts arise as ductal dilatations, usually in the periphery of the kidney, and they communicate (51). Metaplastic cartilage, when present, is also located peripherally, but cartilage is not always present. Some multicystic kidneys contain masses of undifferentiated cells, referred to as nodular blastema (50,52–54). Nodular blastema carries a risk of neoplasia, and it may be related to the rare development of Wilms tumors in multicystic kidneys. Renal cell carcinoma has also been described as a complication of multicystic kidney (55,56). Multicystic dysplastic kidney is almost always associated with ureteral atresia and pyelocalyceal occlusion (57), and the ureter may be partially absent (Fig. 40.4) (58,59). The presence of patent pelvis and calyces in a cystic dysplastic kidney indicates some other type of dysplasia. Cases described in the literature as hydronephrotic multicystic kidney (60), with low ureteral atresia and dilated pelves, are probably variants of obstructive dysplasia and congenital hydronephrosis. Aplastic kidneys also have atretic ureters, but the association is not as clear because several types of small dysplastic kidneys have been grouped together under that heading. Kidneys with atretic ureters do not get infected and are not subject to reflux (61); the occurrence of chronic pyelonephritis in a predominantly dysplastic kidney indicates lower urinary tract obstruction with probable vesicoureteric reflux.
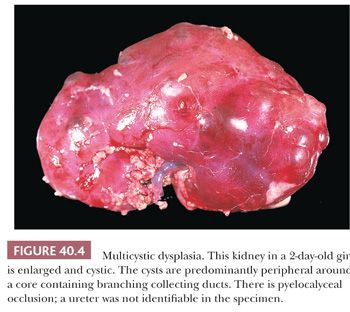
Multicystic and aplastic kidneys are nonfunctional, even though the multicystic kidney may concentrate contrast medium during high-dose excretory urography (62,63) or radionuclide during renal scans (64). Multicystic kidneys are in a dynamic state in early life, and serial ultrasound examinations have shown that the cysts change in size and assume different configurations (65), and they may regress postnatally (66–69). The overall reported incidence of MCKD is between 0.3 and 1 in 1000 live births, with more than half of the cases detected antenatally (70). MCKD may involve both kidneys but is most often unilateral, with the left kidney being more often affected. The incidence is greater in boys than in girls. In the majority of cases, involution of the dysplastic kidney occurs as demonstrated by repeat ultrasound examination, with the rate of involution being the greatest during the first 2 to 3 years of life (71,72).
Deregulation of cell survival in cystic and dysplastic renal development may be related to fulminant apoptosis that occurs in null mutations of bcl–2, an oncogene that suppresses apoptosis, and may contribute to the progressive destruction of functional kidney tissue in cystic kidneys and the spontaneous involution reported in cystic dysplastic kidneys (73,74).
The multicystic kidney is usually detected in the newborn as a flank mass; sonography shows large, spherical cysts with nondelineation of the renal sinus (75,76). Studies have shown that activated p38 and extracellular signal–regulated kinase (ERK) may mediate hyperproliferation and dysplastic tubules, resulting in cyst formation (77). There is a high frequency of contralateral renal and urinary tract abnormalities (78–80), perhaps as high as 40% (81). Malformations of other systems are common, especially congenital heart disease and esophageal or intestinal atresia. Multicystic kidneys are usually unilateral, occasionally bilateral.
Until recently, routine treatment was nephrectomy, partly because of diagnostic uncertainty and partly because of the fear of neoplasia. There is a trend toward conservative management (71,72,82,83), with careful follow-up for any suspicious change in status. That approach, however, is not without controversy (84,85). An indication for nephrectomy is hypertension, which typically resolves after surgery (86,87).
Renal dysplasia may accompany almost any type of congenital urinary tract obstruction, including urethral atresia, posterior urethral valves, anterior urethral diverticulum, and prune belly syndrome (Fig. 40.5). A similar pattern of subcapsular cystic renal dysplasia occurs in some patients with congenital hydronephrosis caused by ureteral obstruction. The renal abnormality takes the form of deficient medullary development and of cystic cortical dysplasia (17,88). The renal cysts in dysplasia associated with lower tract obstruction are usually smaller than those in multicystic kidneys (89). The cortex may contain nests of metaplastic cartilage, sometimes nodules of blastema. The cysts arise, for the most part, in peripheral primitive collecting ducts and collecting tubules, which are admixed with rudimentary glomeruli and convoluted tubules. The abnormal glomeruli and convoluted tubules result partly from arrested development, mostly from involutional and regressive changes in partially formed structures (17). The changes in both cortex and medulla appear to result from the effect of urinary obstruction on development of the outer cortex after nephrons in the inner cortex have begun to excrete urine. The cortical and medullary changes run a spectrum from very mild to extremely severe, and the abnormality is frequently asymmetric, with more frequent involvement of the left kidney. The key to that discrepancy, even in relatively severe congenital bladder outlet obstruction, may be asymmetric vesicoureteric reflux, typically more severe on the left than on the right (90,91). Contralateral vesicoureteric reflux is a complication of unilateral multicystic dysplastic kidney (92–94). Complete bladder outlet obstruction caused by urethral atresia is, on the other hand, almost always associated with bilateral renal dysplasia (85).
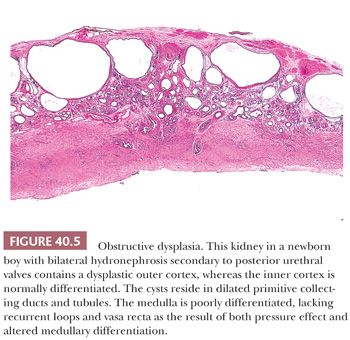
Because obstructive uropathies can be diagnosed before birth by ultrasonography (95), intrauterine surgical relief of the urinary tract obstruction has been tried but remains highly controversial (96). The clinical picture and the treatment are complicated by a high frequency of nonurinary tract malformations (97). Renal failure develops very early in some boys with posterior urethral valves, whereas others, presumably those with less severe obstruction, have minimal symptoms into the third decade of life. Obstructed kidneys are easily infected, and therapy includes removal of obstruction and elimination of vesicoureteric reflux.
Another form of obstructive dysplasia occurs in duplex kidneys, in which one of the ureters has an abnormal bladder insertion and terminates in an ectopic ureterocele. Those circumstances, usually affecting the upper pole ureter, are associated with severe obstruction, resulting in dysplasia (98–100). The other pole may be partially obstructed as the ureterocele enlarges; this pole may then become hydronephrotic, although it is otherwise normally formed. At other times, lower pole reflux may be associated with dysplasia of the lower renal segment (100). Nodular renal blastema occurs in some multicystic kidneys and duplicated kidneys with ureteral ectopy (52,101), perhaps conferring the risk of neoplasia, including Wilms tumor and renal cell carcinoma (102).
Renal dysplasia is encountered in about 10% of refluxing kidneys (103). The dysplastic changes in these circumstances are usually focal, consisting of clusters of primitive ducts and sometimes of cartilage bars, either in scarred and atrophic segments or adjacent to them (Fig. 40.2). The presence of dysplasia is interpreted as the result of intrauterine reflux during metanephric development rather than as acquired postnatal change.
Some dysplastic kidneys, like those in obstructive uropathy, retain limited functional capabilities and have unobstructed urinary systems (48). The patent ureter may be partially obstructed, or dilated or atrophic, but it has the potential for vesicoureteric reflux and ascending infection, with the complications of pyelonephritis and secondary nephrolithiasis. These dysplastic kidneys are difficult to categorize into a single morphologic pattern. Therefore, they lack a generally accepted terminology, and the term hypoplastic dysplasia has been suggested to convey the idea that they are small and partially functional. They have pelves and calyces, which are sometimes dilated, and lobar development is highly variable, with thin cortices (Fig. 40.3). Evaluation of both biopsy and nephrectomy specimens leads to recognition of dysplastic elements in the form of cartilage, primitive ducts, and rudimentary medullary pyramids.
Large and diffusely cystic dysplastic kidneys with patent urinary tracts occur principally in malformation syndromes, occasionally as isolated malformations. They are not to be confused with multicystic kidneys because the clinical and genetic implications are considerably different. The kidneys superficially resemble those of autosomal recessive “infantile” polycystic kidney disease because they have the same reniform external configuration and a finely cystic external surface. Careful gross examination of the sectioned kidney, however, discloses a pattern of rounded—rather than elongated—cysts, and the medullary pyramids and calyces are severely underdeveloped. The cysts in diffuse cystic dysplasia arise principally within primitive collecting ducts, although portions of the nephron also become cystic, as in Meckel syndrome (104). In many specimens, however, there is a striking paucity of nephrons. Some clusters of glomeruli and convoluted tubules are present among the cysts, but normal cortical organization into medullary rays and cortical labyrinth is usually obscured. The association of Dandy-Walker malformation and cystic dysplastic kidneys, as well as occipital encephalocele and polydactyly, may represent pleiotropy/heterogeneity (105); however, familial renal hepatic-pancreatic dysplasia and Dandy-Walker cyst appear to be a separate syndrome. Likewise, Dandy-Walker malformation, cystic renal dysplasia, and hepatic fibrosis may be within the phenotypic expression of Meckel syndrome or a distinct syndrome. Cartilage is seldom present. Diffuse cystic dysplasia occurs regularly in Meckel syndrome (106,107); it occurs less often in a group of disorders that includes several forms of short-limbed chondrodysplasia, Zellweger syndrome, glutaric aciduria type 2, and renal hepatic-pancreatic dysplasia (108). In all of these syndromes, the liver contains a biliary abnormality similar to that of autosomal recessive polycystic kidney disease and congenital hepatic fibrosis. Specific diagnosis depends on recognition of the syndrome because the renal abnormality is similar in all of them.
An important clinical question regarding renal dysplasia concerns the risk of recurrence. Dysplasia, unlike polycystic kidney disease, may be truly unilateral, and it does not eventually involve a previously normal opposite kidney. The risk of inheritance of nonsyndromal dysplasia is small, empirically not significantly different from zero (109). Nonetheless, there is minimal risk of recurrence of multicystic and aplastic kidneys in subsequent siblings in that both malformations occur in the hereditary renal adysplasia syndrome, which comprises unilateral dysplasia, unilateral agenesis, and lethal bilateral agenesis, usually in a dominant pattern of inheritance (110–113). A balanced translocation in one case of bilateral multicystic dysplasia has been reported. There is also a small risk of recurrence in obstructive dysplasia secondary to posterior urethral valves because the valves have a low familial incidence (114). The risk of recurrence is much greater in diffuse cystic dysplasia, in which the risks are those of the associated syndrome.
Multicystic kidneys, which are often diagnosed by prenatal sonography, undergo an evolution to shrinkage and even disappearance (115). The kidney occasionally enlarges (a fluctuation attributable to cyst enlargement), but the general trend toward shrinkage often leads to the ultimate equivalent of agenesis (115).
RENAL TUBULAR DYSGENESIS
An unusual cause of neonatal oliguria, usually after a gestation complicated by oligohydramnios, is renal tubular dysgenesis (RTD) (116–118). Oligohydramnios may be delayed, however, becoming evident after the 20th week of gestation (119), which makes early diagnosis difficult even when the condition is suggested by family history. The renal abnormality has been associated with widely patent cranial fontanels (119,120). The kidneys are commonly, although not necessarily, enlarged, containing an increased number of nephrons (117). The cortical tubules are lined with densely packed columnar cells that histochemically bind peanut lectin, and the immunohistochemical (IHC) reaction for epithelial membrane antigen is positive, suggesting that the tubular segments are of collecting duct origin (Fig. 40.6). The glomeruli appear to be crowded together and the medullary pyramids are smaller than normal.

IHC demonstration of very large amounts of renin within preglomerular arteries suggests vasoconstriction and greatly reduced glomerular perfusion (121). A microdissection study has demonstrated marked shortening of all the nephron segments from the glomeruli to the collecting tubules rather than an isolated abnormality of the proximal convoluted tubules (122).
Recognition of this condition is of great importance for family counseling because it has been shown to have an autosomal recessive inheritance (119). The renal abnormality has also been linked to treatment of maternal hypertension with angiotensin-converting enzyme (ACE) inhibitors (123,124) and to maternal use of cocaine (118,125,126).
A renal abnormality bearing some similarities to that found in familial RTD occurs after maternal use of nonsteroidal anti-inflammatory drugs (NSAIDs). The same effect on fetal urine output and amniotic fluid volume is common to all NSAIDs, and the risk of fetal renal injury may be common to all (127). Abnormal fetal renal tubular development and oligohydramnios have been attributed to treatment of pregnant women with indomethacin (128), a drug used as a tocolytic agent and inhibitor of fetal urine output. Renal failure dysgenesis with hypocalvaria and twin-to-twin transfusion syndrome has been attributed to indomethacin treatment in the mother for acute polyhydramnios (129). Fetuses in twin pregnancies with polyhydramnios are at increased risk of NSAID-associated renal injury.
RTD should be obvious by the 30th week of gestation. Metzman et al. (130) have described partial tubular differentiation at 20 and 22 weeks, with the suggestion that regressive changes contribute to the pathogenesis of RTD.
Late second trimester demonstration of oligohydramnios with structurally normal kidneys and with or without skull ossification defects allows the diagnosis of RTD, which, however, should be confirmed by histologic and immunohistologic examinations of the kidney (131).
Ischemia may play a role in RTD. Reduced renal blood flow, whether from primary renin-angiotensin abnormalities or ACE-inhibitor–mediated fetal hypotension, may contribute to poor tubular growth and differentiation, as suggested by Landing et al. (132), and lack of angiotensin II as growth factor may play a key role in both the familial and ACE-inhibitor–induced RTD (132). RTD has been reported in association with meconium ileus in neonatal hemosiderotic liver disease.
POLYCYSTIC KIDNEY DISEASE
Polycystic disease of the kidney in children comprises two genetically different disorders: one with autosomal recessive inheritance and an onset typically in childhood, and the other with an autosomal dominant inheritance and an onset occasionally in childhood. The clinical, morphologic, and radiographic characteristics overlap, and differentiation may become apparent only on extensive investigation of the family. Molecular genetic testing for both autosomal recessive as well as autosomal dominant polycystic kidney disease is available and is increasingly utilized given the high mutation detection rates (133). Nonetheless, the distinction between these disorders is important for prognostication and family counseling. Renal cystic disease also occurs in several hereditary syndromes, particularly tuberous sclerosis. Glomerulocystic disease is a morphologic term descriptive of the renal abnormality occurring in some cases of childhood autosomal dominant polycystic kidney disease, tuberous sclerosis, and certain other syndromes. Some of these cystic diseases can be encountered by the surgical pathologist in renal biopsy specimens. A classification of renal cystic diseases is listed in Table 40.1.
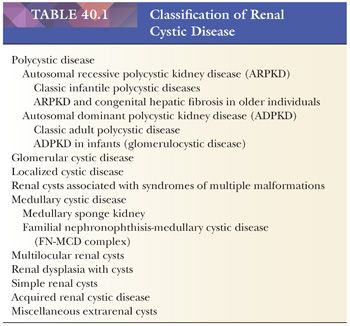
AUTOSOMAL RECESSIVE POLYCYSTIC KIDNEY DISEASE
Autosomal recessive polycystic kidney disease (ARPKD) is a rare condition with an incidence of 1 in 6000 to 1 in 14,000 births. The gene has been mapped to the short arm of chromosome 6. The locus PKHD1 (polycystic kidney and hepatic disease 1) on chromosome 6p21.1 has been linked to all classic forms of this disorder (134). It may consist of several clinically and morphologically overlapping syndromes (135), each with autosomal recessive inheritance, or it may be a single disorder subject to modification by gene linkages and epigenetic factors (108). In this discussion, ARPKD is considered as one genetic disorder, with different phenotypes (136) that, in a sense, relate to differences in severity of expression. There is no confirmed evidence of genetic heterogeneity. Newborn infants typically have nonfunctioning, enlarged, diffusely cystic kidneys; the livers contain enlarged portal areas with apparent biliary proliferation. Older infants have less severely enlarged kidneys, with less functional impairment and less severe cyst formation, and the same hepatic abnormality. Some older children and adults have asymptomatic renal involvement, with very few cysts, and progressive portal fibrosis, known as congenital hepatic fibrosis (137), leading to portal hypertension. Although certain generalizations can be stated, sharp distinctions among these groups cannot be made. More than one pattern has occurred among affected siblings in the same family (138–141). The molecular pathogenesis of ARPKD has been identified as PKHD1 gene product, a novel protein, fibrocystin (142). Despite the identification of PKHD1 and its protein product, fibrocystin, the pathogenesis of ARPKD remains unclear (143–145). Reduced or absent function of fibrocystin is thought to underlie the disease pathogenesis (146,147). Recent studies suggest that many polycystic kidney disease–related proteins are involved with the function of primary cilia, an organelle located on the apical surface of most epithelial cells including kidney tubule and biliary cells (148,149). Abnormal structure and/or function of the primary cilium lead to alterations in its mechanosensory properties, which may result in activation of downstream second messenger pathways, which in turn are thought to activate known cystogenic processes such as cell proliferation and fluid secretion (149,150).
Newborns with the severe form of the disease die shortly after birth, usually because of respiratory insufficiency. Many of them have the Potter sequence, with its characteristic facies, secondary to oligohydramnios, and pulmonary hypoplasia, which leads to the postnatal complications of severe respiratory distress. The kidneys are often huge, together exceeding 300 g. They have a reniform configuration and a normal lobar structure, but they are spongy and externally finely cystic. The cysts are located in collecting ducts and tubules (151–154), which appear to be elongated sacs lined with cuboidal epithelium (Fig. 40.7A). Lectin markers (e.g., peanut lectin) clearly identify cysts as dilated collecting ducts (155); the lectins may also bind to distal tubular segments (104,156). The cystic ducts on the cut surface run from medulla to outer cortex, and the dilated medullary ducts tend to have a more rounded appearance than do the cortical ducts. The cortex is commonly edematous, accounting for some of the increased weight of the kidney and separating the individual nephrons so they seem to be reduced in number. The medullary pyramids, apart from the cysts, are normally formed and the essential architecture of the kidney thus appears to be normal. The livers contain enlarged portal areas with an increased number of biliary profiles that form an array of anastomosing channels (see Fig. 40.7B). However, the biliary structures are flattened sacs rather than ducts (157–159), and occasional specimens contain gross cysts. Cysts in viscera other than liver and kidney are rare in homogeneous populations of ARPKD, and their presence may be clues to other syndromes.
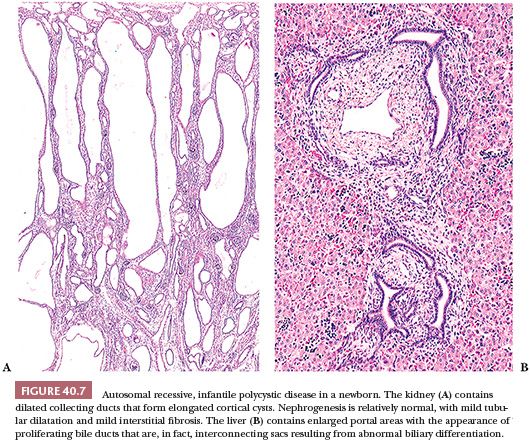
Older infants have more irregular renal cyst formation, and asymptomatic older children may have very few cysts. The cysts are located predominantly in the outer medulla (160). The histopathologic picture becomes complicated, however, because of tubular atrophy and interstitial fibrosis (Fig. 40.8). The cortical cysts often have a rounded appearance, suggesting localized obstruction. Dilated convoluted tubules become mildly cystic, perhaps as the result of secondary local obstruction. Whether the disease in those patients in whom renal insufficiency develops and progresses later in life is due to additional cyst formation (161) or by tubular atrophy and interstitial fibrosis (159) is uncertain, although the former appears to be uncommon. The hepatic abnormality, although essentially the same as that seen in infants, becomes increasingly fibrotic with time. Perilobular fibrosis and regenerative nodules may result in a pattern resembling micronodular cirrhosis (162). It is thought that portal hypertension results from presinusoidal obstruction, but there may also be an element of postsinusoidal obstruction (163) because central perivenous fibrosis is relatively common (164). Although the abnormal portal biliary passages commonly contain plugs of inspissated bile, hepatocellular changes and intralobular cholestasis do not usually occur except in the presence of superimposed infection. Ascending suppurative cholangitis is a relatively common complication (162,165), probably related to biliary dilatation and stasis. Nonobstructive ectasia of the intrahepatic ducts, known as Caroli disease, occurs in a small proportion (perhaps 10%) of patients with ARPKD (162,166,167). The same or very similar intrahepatic biliary abnormalities are seen in conjunction with other hereditary syndromes with renal cystic or dysplastic changes, notably the Meckel, Jeune, and Zellweger syndromes. It has also been described in some patients with juvenile nephronophthisis. All these conditions, like ARPKD, are inherited as autosomal recessive traits.
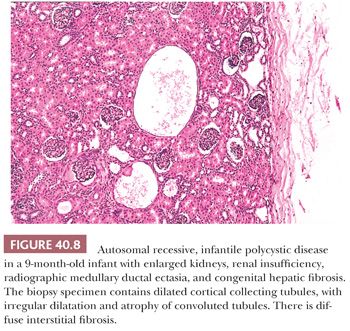
The histopathologic diagnosis of ARPKD in a newborn with typical, severe renal involvement is usually straightforward. The abnormality must be differentiated, as discussed previously, from diffuse cystic dysplasia, which has different morphologic characteristics. The diagnosis often can be confirmed by a liver biopsy, but open-wedge biopsies are preferable to percutaneous needle biopsies for this purpose. However, the histopathologic diagnosis of the renal abnormality in older children can be difficult, as attested to by some confusion in the literature and by difficulties in evaluating individual cases. The hallmark of ARPKD in the older child is medullary ductal ectasia (168), but urography has been supplanted by sonography, which shows variably enlarged kidneys with increased echogenicity (169). The sonographic appearances are similar to those of autosomal dominant polycystic kidney disease in childhood and of glomerulocystic kidney disease. Liver biopsies in older children may also be helpful by showing the characteristic biliary abnormality.
Older patients have variable progression of the renal abnormality (170). In some, renal insufficiency develops in early childhood, and others remain asymptomatic into adulthood. Patients typically have a concentrating defect and impaired acidification unrelated to reduced glomerular filtration. Systemic hypertension develops in almost all patients, and cerebral arterial aneurysms, although less common than in patients with autosomal dominant polycystic kidney disease, do occur. Portal hypertension and its complications also develop in older children and young adults. Some patients have both renal insufficiency and portal hypertension, whereas others have only mild urinary concentrating defects. Clinical evidence of hepatocellular dysfunction may develop in relation to biliary infection, but patients are ordinarily not jaundiced.
Differential Diagnosis
The differential diagnosis in older children includes other causes of progressive renal insufficiency with a concentrating defect, the most common of which is familial juvenile nephronophthisis, a form of hereditary interstitial nephritis. That abnormality is often accompanied by medullary cysts, and the kidneys are very small because of severe cortical atrophy. Urographic studies do not, therefore, visualize the medullary cysts, and sonographic studies may be equivocal. A small proportion of patients with nephronophthisis have had hepatic involvement in the form of portal fibrosis. More commonly associated abnormalities include pigmentary retinal degeneration (renal-retinal dysplasia) and skeletal dysplasia.
Fetal ultrasonography offers the possibility of prenatal diagnosis of ARPKD. The fetal kidneys are enlarged bilaterally and have increased echogenicity. Ultrasonic diagnosis is straightforward when other siblings are known to be affected, but, in an index case, distinction from other forms of cystic disease, such as diffuse cystic dysplasia or glomerulocystic disease, may be difficult.
In a child with clinically enlarged kidneys that appear diffusely hyperechoic on ultrasound, the appearances on Tc-99m dimercaptosuccinic acid (DMSA) imaging strongly support the diagnosis of ARPKD. The Tc-99m hepatic iminodiacetic acid (HIDA) findings, especially of an enlarged left lobe of the liver with bile stasis or dilatation, further support the diagnosis (171).
AUTOSOMAL DOMINANT POLYCYSTIC DISEASE
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited human kidney disease with an incidence of approximately 1:400 to 1:1000. It affects all races and both sexes equally, although it may be more severe in males (172). ADPKD occasionally has an early onset in the newborn or young infant, sometimes before the onset of clinically apparent renal disease in a parent (153,173–176). Morphologic studies of the affected infants have shown the relatively frequent occurrence of glomerular cysts (159,177,178) to an extent that so-called glomerulocystic kidney disease (GCKD) is a common expression of ADPKD in very young children. Several studies of affected parent and fetus by DNA-probe analysis have shown the same linkage to chromosome 16 as in classic ADPKD (179–181). In GCKD, parental ultrasound should be performed to exclude ADPKD; neonatal GCKD may antedate the appearance of classic ADPKD in one of the parents.
ADPKD is genetically heterogeneous (182). Two major disease-causing genes are PKD1 and PKD2. PKD1 has been mapped to chromosome 16 and accounts for nearly 85% of cases. The remaining cases are associated with PKD2 that is linked to chromosome 4q13-q23 (183). A third locus has also been suggested (184). Protein products of PKD1and PKD2 are called polycystin-1 (PC1) and polycystin-2 (PC2) respectively. PC1 and PC2 are thought to interact to form a functional complex. Accumulating evidence indicates that, in keeping with many other polycystic kidney disease proteins, PC1 and PC2 are localized to primary cilia and polycystic disease is a ciliopathy (185,186). Genetic heterogeneity, mutation position in PKD1, mutation type, modifier genes, and environmental factors account for the substantial variability in severity of renal disease and other manifestations of ADPKD (187,188). A clear association exists between the severity of renal disease and the gene involved (PKD1 or PKD2). Mutations in PKD1 are associated with more severe disease, with an earlier age at diagnosis and mean age of onset of ESRD (189).
The kidneys in affected infants are enlarged and hyperechoic, much like the kidneys in typical recessive disease (173,190). Young infants may have renal insufficiency and die of renal failure, but some of them seem to stabilize, with survival into the second or third decade. Very severe cortical cystic disease is accompanied by abnormal medullary development, a form of dysplasia (153,178). In older children identified by clinical screening as having more typical ADPKD with localized cysts, renal insufficiency does not ordinarily develop until adulthood (191). Hepatic involvement, with biliary dysgenesis resembling that of ARPKD, occurs in approximately 10% of young children. Children also may have hypertension and cerebral berry aneurysms.
The kidneys in older children, as in adults, commonly contain irregularly clustered small cysts, and the cysts are lined with hyperplastic epithelium (Fig. 40.9). Light, transmission, electron, and scanning electron microscopic studies of ADPKD kidneys have shown micropapillary epithelial hyperplasia with small intracystic and intraductal polyps, and the epithelial hyperplasia is believed to contribute to cyst formation by causing luminal obstruction (192–195). The cysts arise in collecting ducts and in all portions of the nephron, as shown by lectin-binding studies (155) that confirm older microdissection studies. Only a minority of nephrons, probably less than 20%, become cystic, and the disease progresses by cyst enlargement with compression of parenchyma rather than by recruitment of new cysts (196). Cyst enlargement, with increased renal volume, correlates also with the clinical development of hypertension (197). Cysts often become infected (198,199), and there is a very high frequency of nephrolithiasis (200). Hepatic cysts increase in frequency with age (201) and become infected (202). A few families have had a liver abnormality indistinguishable from congenital hepatic fibrosis in ARPKD (178). Cardiac valvular lesions, intracranial aneurysms, and colonic diverticula also develop (159).

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


