Chapter 13 Degenerative and metabolic diseases
The hyperlipidemias
The last, xanthoma disseminatum, in which serum lipid levels are normal, is discussed in Chapter 29 (see xanthogranuloma). Xanthoma cells express CD4, CD11c, CD14b, and CD68 in addition to human leukocyte antigen (HLA) class II antigens.2
Hyperlipidemias may be primary, or secondary to conditions such as diabetes mellitus, obesity, pancreatitis, renal disease (the nephrotic syndrome or chronic renal failure), hypothyroidism, alcohol consumption, pregnancy, cholestatic liver disease (e.g. primary biliary cirrhosis) and paraproteinemias. Drug-induced hyperlipidemia also occurs as a result of administration of estrogens, corticosteroids or 13-cis-retinoic acid. It is often associated with serious, potentially life-threatening disorders such as atherosclerosis (low density lipoproteins) and pancreatitis (hypertriglyceridemia).3
The presence of xanthomata commonly represents a cutaneous manifestation of systemic disease, and their recognition should therefore be followed by an intensive investigation to exclude the latter (Table 13.1).2,4 Although not a hard and fast rule, xanthoma morphology and distribution can sometimes point towards specific hyperlipidemia variants.
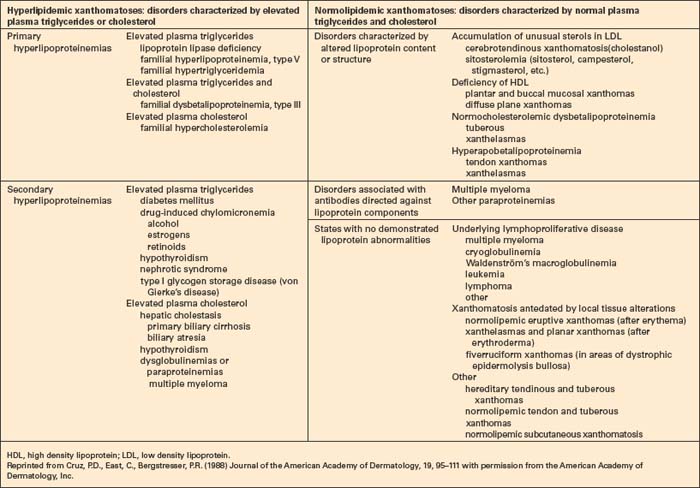
The plasma lipids are composed of triglycerides and cholesterol; these are insoluble and their transport is facilitated by their aggregation into lipoproteins. The latter are macromolecular complexes composed of an outer shell of hydrophilic phospholipids, nonesterified cholesterol and apo(lipo)proteins, which emulsify the associated hydrophobic core of triglycerides and cholesterol ester.5 There are a large number of apoproteins, with variable structure and function (e.g. ApoB-48, which is required for the secretion of chylomicrons into the thoracic duct).3 In addition to giving structure to the lipoprotein, apoproteins also represent ligands for specific receptors (e.g. ApoE is a ligand for liver chylomicron receptors). They also act as cofactors for a number of lipid-modifying enzymes (e.g. ApoCII activates lipoprotein lipase).5 Lipoprotein metabolism, which is summarized in Figure 13.1, involves both exogenous (dietary) and endogenous pathways.6 For more detailed information the reader is particularly referred to references 1 and 6.
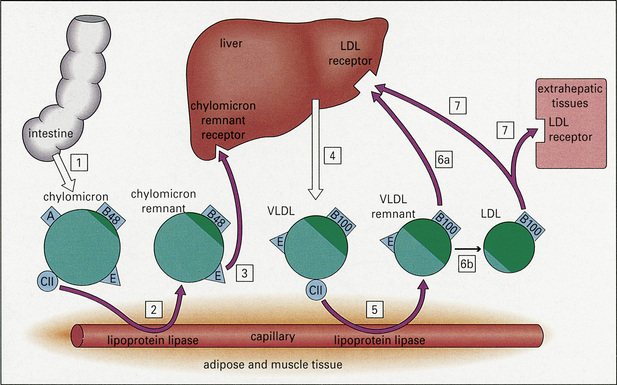
Fig. 13.1 Lipoprotein metabolism. (LDL, low density lipoprotein; VLDL, very low density lipoprotein.)
Reproduced with permission from Cruz, P.D., East, C., Bergstresser, P.R. (1988) Journal of the American Academy of Dermatology, 19, 95–111.
The classification of hyperlipidemias is based upon the electrophoretic separation, on paper or agarose gel, of abnormal quantities of lipoprotein in the plasma (Fig. 13.2). There are seven main classes of lipoprotein, with differing electrophoretic mobilities:
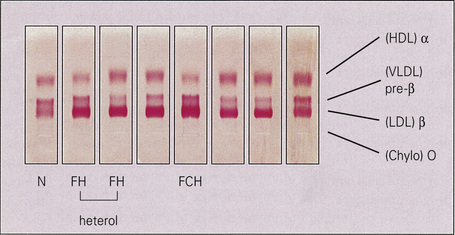
The hyperlipidemias are classified into six types according to the lipoprotein anomaly present (Table 13.2). However, it should be noted that each of these six types may result from a variety of pathogeneses, including those of a known or presumed genetic basis and others that complicate a diverse group of disease processes (secondary hyperlipidemia).7–9 High density lipoproteins are not atherogenic.3 Indeed, their function is to remove cholesterol from the tissues and high levels serve to protect against vascular disease.5 Conversely, HDL deficiency (e.g. Tangier disease) is associated with cholesterol accumulation.10
The lipid content of xanthomata is probably mostly derived from the plasma, presumably by lipoprotein (particularly LDL and VLDL) permeation of blood vessel walls with the release of lipid and its subsequent phagocytosis by histiocytes, although localized lipogenesis may also be of importance.10–13 The subgroups and proportions of lipid deposited within xanthomata are similar to those found in atheromatous plaques, raising the possibility of a shared pathogenesis.1
Xanthomata are, however, not always associated with hypercholesterolemia or hyperlipoproteinemia.14 Under such circumstances, they may evolve as a consequence of altered lipoprotein content or structure, represent local tissue changes or develop as a consequence of systemic disease including lymphoma, multiple myeloma, and Waldenström’s macroglobulinemia.15 Normocholesterolemic xanthomata can therefore arise as a consequence of the accumulation of cholesterol-like substances within histiocytes (e.g. cerebrotendinous xanthomatosis and β-sitosterolemia).
Cerebrotendinous xanthomatosis represents an abnormality of bile acid metabolism inherited in an autosomal recessive pattern.16–18 As a consequence of mitochondrial enzyme sterol 27-hydroxylase deficiency and resultant impaired oxidation of the cholesterol side chain during the production of cholic acid, cholestanol (and cholesterol) accumulates in the tissues, especially the tendons, lungs, and brain. The xanthomata particularly affect the Achilles tendons and the tendons of the knees, elbows, and the interphalangeal joints.19 In addition to tendinous xanthomata, patients develop juvenile cataracts and progressive neurological dysfunction including mental retardation, dementia, pyramidal signs, cerebellar ataxia, spinal cord paresis, and sensory changes due to dysmyelination.18,20,21 Coronary atherosclerosis, endocrine abnormalities, and diarrhea may also be present. In addition to cholestanol accumulation, cerebrotendinous xanthomatosis has been shown to be characterized by abnormal high density lipoproteins, which result in impaired cholesterol (and cholestanol) transport and contribute to the consequent xanthomatization.16 The mortality is high, patients usually dying in the fourth to sixth decades, most often from progressive neurological dysfunction, pseudobulbar paralysis or myocardial infarction.21
Tendinous and tuberous xanthomata may also represent a manifestation of β-sitosterolemia. This is an autosomal recessive condition in which increased intestinal absorption of the plant sterols β-sitosterol, campesterol, and stigmasterol results in tissue deposition along with cholesterol and subsequent xanthoma formation.22–24 Normally these sterols are almost completely unabsorbed from the gastrointestinal tract. β-Sitosterolemia is associated with an increased risk of atherosclerosis.3
Xanthomata may occur in extracutaneous locations mimicking tumors in patients with hyperlipidemia. Sites include deep soft tissues and mediastinum.25
Eruptive xanthomata
Clinical features
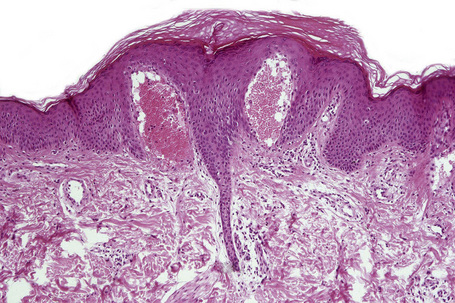
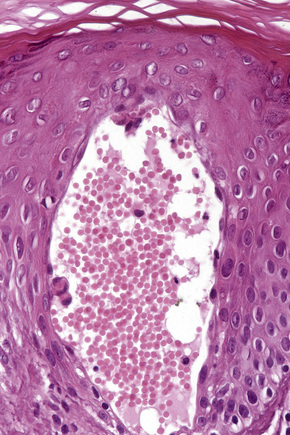
Eruptive xanthomata are small (1–4 mm) yellowish papules with a red halo that have a predilection for the buttocks, shoulders, and extensor surfaces of the limbs (Fig. 13.3).1 They may also present in the antecubital and popliteal fossae, axillae, lips, eyelids, and ears.2 They often appear in crops and may wax and wane with plasma lipoprotein levels.3 Lesions usually resolve spontaneously over a period of weeks. Pruritus is frequently present and the papules are sometimes tender.2 Eruptive xanthomata may rarely display a Koebner phenomenon.4,5 Healing is occasionally associated with the development of hyperpigmented scars.2 Cutaneous lesions of Langerhans cell histiocytosis may mimic eruptive xanthoma.6
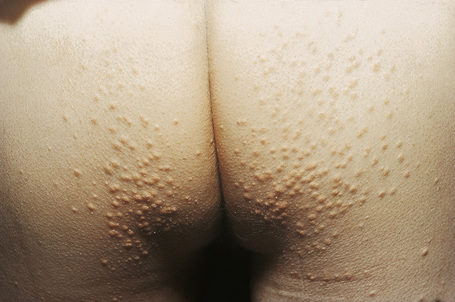
Fig. 13.3 Eruptive xanthoma: numerous small yellow papules are present on the buttocks.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
Eruptive xanthomata are associated with hypertriglyceridemia and most often occur in hyperchylomicronemic states. Sometimes their presence correlates with increased levels of very low density lipoproteins. The most common cause, however, is secondary hyperlipoproteinemia, especially in those cases associated with diabetes mellitus and alcohol ingestion, or in those that are drug induced (e.g. due to exogenous estrogens, corticosteroids or retinoids).2,7 They may also develop as a consequence of decreased lipoprotein lipase activity, ApoCII deficiency or increased synthesis of VLDL, which effectively blocks chylomicron access to lipoprotein lipase.2,8 Eruptive xanthomata are therefore often accompanied by other features of hyperlipidemia, including lipemia retinalis, hepatosplenomegaly, abdominal pain, and pancreatitis. They may also rarely develop as a manifestation of primary hyperlipoproteinemia (HPL), particularly autosomal recessive lipoprotein lipase deficiency (HLP type I) in children and familial HPL type V in adults.9,10 An exceptional association with β-sitosterolemia, a condition usually presenting with tuberous or tendinous xanthomata, has been documented.11 Much rarer associations include familial hypertriglyceridemia, the nephrotic syndrome, chronic pancreatitis, von Gierke’s disease, and hypothyroidism.7,12,13 An association with acanthosis nigricans has also been reported.14
Histological features
The histological features are seen predominantly within the superficial reticular dermis. In early lesions histiocytes are numerous and the fully developed ‘foam cells’, which characterize xanthomata, are sometimes few in number. The infiltrate may also contain an admixture of lymphocytes and neutrophils.15,16 In an established papule, xanthoma cells with characteristic clear or foamy cytoplasm form the predominant cell type (Figs 13.4–13.6).
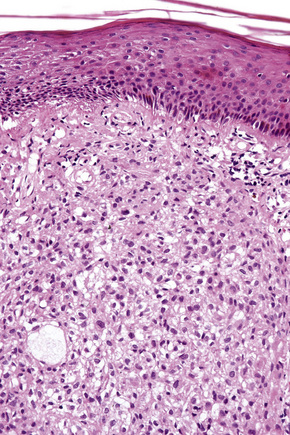
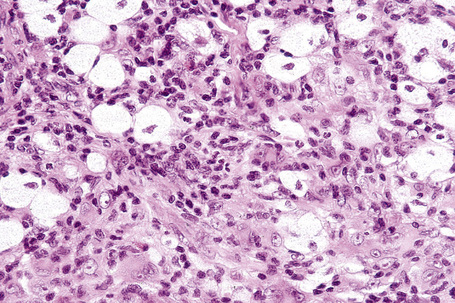
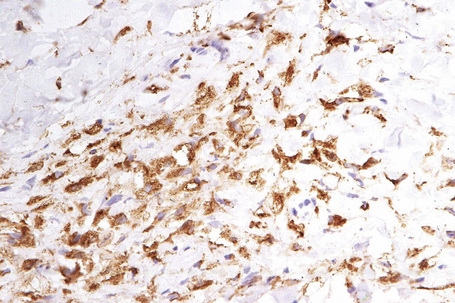
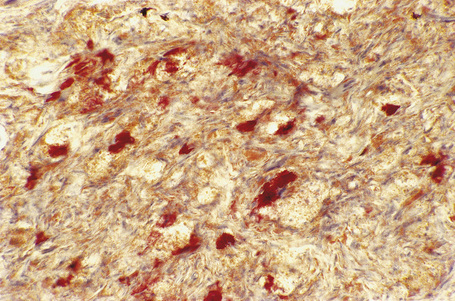
Eruptive xanthomata often develop rapidly over the course of several days and occasionally are associated with spontaneous resolution. The quantity of intracytoplasmic lipid (predominantly triglyceride in contrast to other xanthomata, which contain mostly cholesterol) is in a state of flux and may be associated with extracellular deposition, a phenomenon that is rare or absent in the other types of xanthomata. In all xanthomata the lipid within the macrophage stains positively with fat stains such as oil red O, scarlet or Sudan red (Fig. 13.7).
Differential diagnosis
There can be confusion with granuloma annulare histologically as both conditions have certain features in common, namely a dermal interstitial histiocytic infiltrate with variably increased mucin.12,17 Although extracellular lipid may disrupt dermal collagen, necrobiosis is not characteristic of eruptive xanthoma. Additionally, it contains few giant cells and the perivascular infiltrate is lymphocytic, in contrast to the histiocytes seen in granuloma annulare.12
Tendinous xanthomata
Clinical features
Tendinous xanthomata, which are associated with raised low density lipoprotein levels, are slowly enlarging subcutaneous tumors that occur in tendons (especially those of the hands, knees, elbows, and the Achilles tendon), ligaments, fascia, and periosteum (Figs 13.8, 13.9).1 The overlying skin, which appears normal, is freely moveable over the surface and small tendon xanthomata may be difficult to palpate.1 The lesions characteristically ‘move with the tendons’ and are thought to be trauma related.2 The presence of these xanthomata is most frequently a feature of heterozygous familial (LDL receptor deficiency) hypercholesterolemia.2–4 There is a high risk of associated coronary atherosclerosis. A recent meta-analysis demonstrated a threefold increased risk of cardiovascular disease in patients with familial hypercholesterolemia and tendinous xanthomata compared to those without cutaneous lesions.5 Tendinous xanthomata are also seen in familial combined hyperlipidemia, normocholesterolemic states such as cerebrotendinous xanthomatosis (cholestanolosis) and β-sitosterolemia, and the nephrotic syndrome.2,6–10
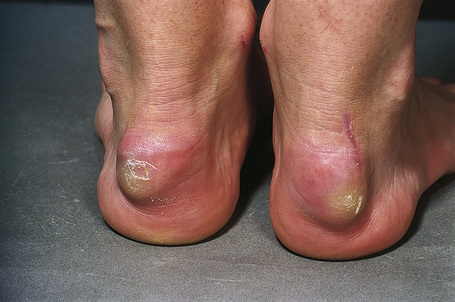
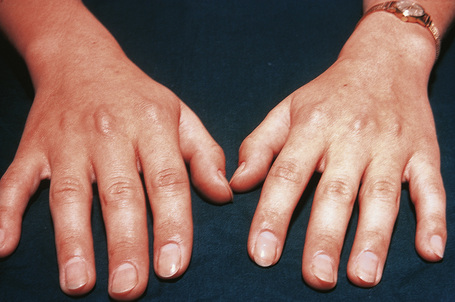
Fig. 13.9 Tendinous xanthoma: xanthomata are present overlying the knuckles.
By courtesy of the Institute of Dermatology, London, UK.
Histological features
Tendinous xanthomata are composed of multiple nodules containing xanthoma cells, accompanied in early lesions by an admixture of inflammatory cells including histiocytes, lymphocytes, and neutrophil polymorphs. The deposits in tendinous xanthoma are doubly refractile to polarized light (Fig. 13.10). Older lesions are characteristically associated with fibrosis.

Tuberous xanthomata
Clinical features
Tuberous xanthomata are firm yellow–red papules and nodules, which are found most frequently on the extensor aspect of the knees, elbows, and buttocks (Figs 13.11–13.13).1 Lesions sometimes also occur on the hands and palms.2 They are most characteristically seen in familial dysbetalipoproteinemia type III, and there is a particular risk of peripheral vascular disease.3 Four other conditions may also be characterized by tuberous xanthomatosis:
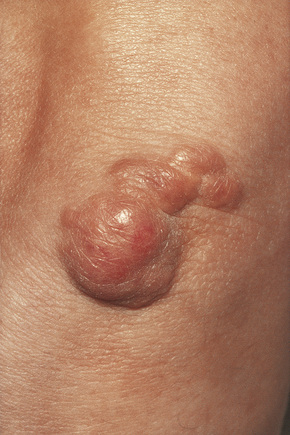
Fig. 13.11 Tuberous xanthoma: firm erythematous nodules over the elbow.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
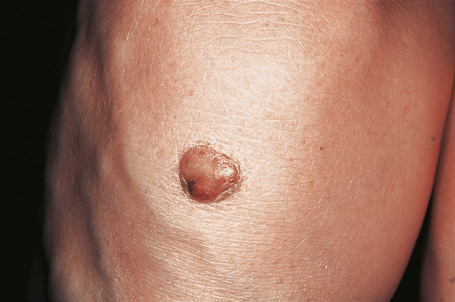
Fig. 13.12 Tuberous xanthoma: erythematous nodule on the back of the arm.
By courtesy of the Institute of Dermatology, London, UK.
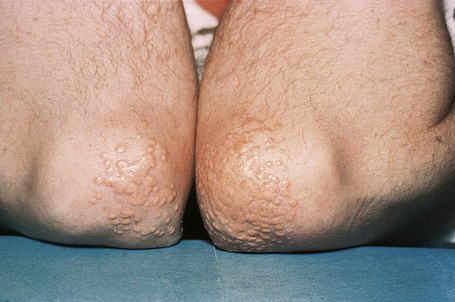
Fig. 13.13 Tuberous xanthoma: in this example, eruptive lesions are present on the elbows.
By courtesy of the Institute of Dermatology, London, UK.
Tuberous xanthomata also occur in secondary hyperlipidemia (e.g. due to the nephrotic syndrome or hypothyroidism). Protease inhibitors may cause hyperlipidemia, and ritonavir has been reported to induce tuberous and tendinous xanthoma lesions.5 Clinically, tuberous xanthomata occasionally resemble the lesions of erythema elevatum diutinum. Tuberous and tendinous normolipemic xanthomata have been described but it seems that, with adequate follow-up, patients usually develop some form of hyperlipidemia.6 Cholesterotic fibrous histiocytomas may be associated with hyperlipidemia and often simulate a tuberous xanthoma clinically and histologically.7 A rare case of malignant fibrous histiocytoma clinically presenting as a tuberous xanthoma in a patient with type IIA hyperlipoproteinemia has been documented.8
Histological features
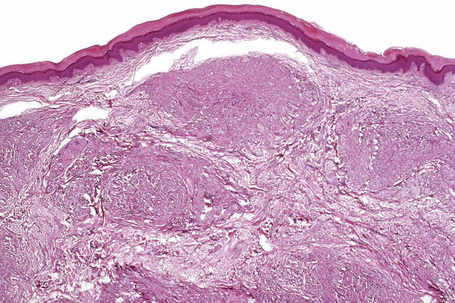
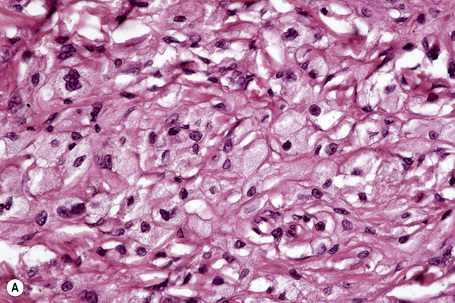
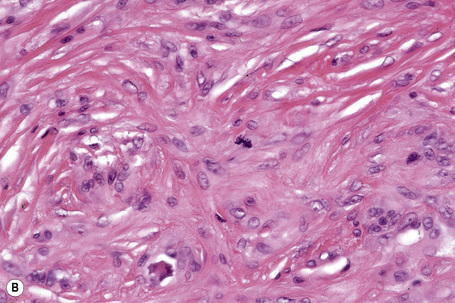
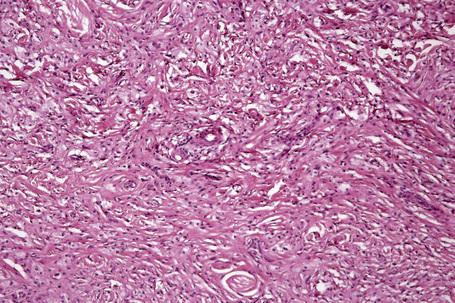
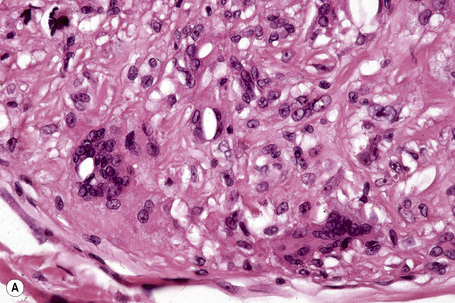
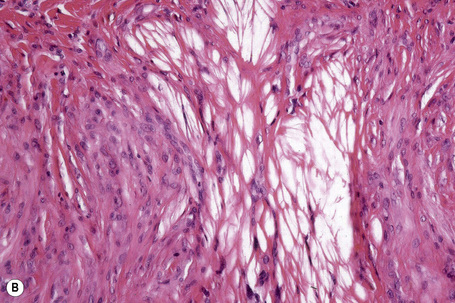
Tuberous xanthomata consist of multiple nodules in the reticular dermis and sometimes the subcutaneous fat (Fig. 13.14). Their appearance varies, depending upon their stage of evolution (Fig. 13.15). Xanthoma cells predominate in early lesions, but with maturity fibrosis supervenes (Fig. 13.16). On occasion, foreign body giant cell granulomata containing cholesterol clefts are seen and a perivascular chronic inflammatory cell infiltrate is sometimes evident (Fig. 13.17).
Planar xanthomata
Clinical features
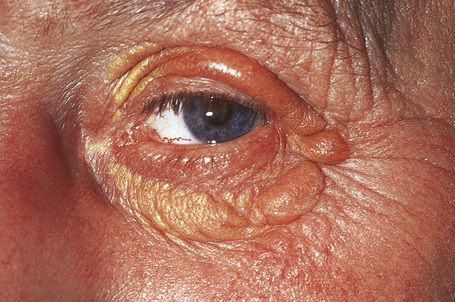
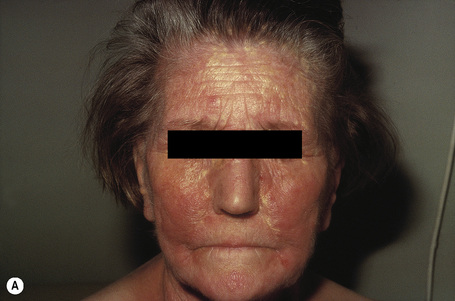
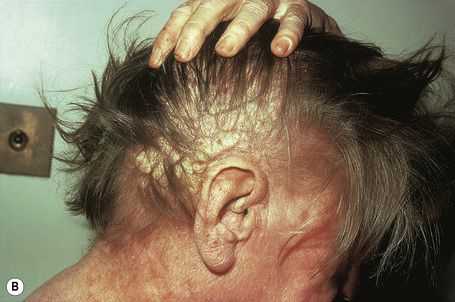
Planar xanthomata are typically soft yellow dermal macules or plaques that occur most frequently around the eyes, where they are known as xanthelasmata (Fig. 13.18).1,2 About 50% of patients with xanthelasmata have associated hyperlipidemia (hypercholesterolemia or hyperlipoproteinemia type III) which is often accompanied by a cholesterol corneal arcus.3–5 Many of those who appear biochemically normal on routine testing, however, are shown to have subtle abnormalities of lipid metabolism on more detailed analysis.6 There is a particularly increased risk of coronary artery atherosclerosis in younger patients.1 When very extensive (diffuse or generalized plane xanthomatosis) and associated with orange–yellow planar xanthomata around the head and neck, and occasionally the upper trunk and arms, there may be an associated systemic disorder such as multiple myeloma with paraproteinemia, cryoglobulinemia, benign paraproteinemia or, less commonly, leukemia and rheumatoid arthritis (necrobiotic xanthogranuloma) (Fig. 13.19).1,3,7–13 More exceptional associations include idiopathic Bence Jones proteinuria, Sézary’s syndrome, Castleman’s disease, relapsing polychondritis, acquired palmoplantar keratoderma, adult T-cell leukemia/lymphoma, and Takayasu’s disease.14–20 A patient with monoclonal gammopathy and cutaneous lesions with features of both plane xanthoma and amyloidosis has been documented.21 The latter case was also associated with myeloma.
In cases of myeloma and plane xanthoma, it has been demonstrated that complexes form between serum lipoproteins and paraprotein, suggesting that this interaction may induce a hyperlipidemia and xanthoma formation.22,23 The serum lipid levels of patients with diffuse plane xanthomata are normal or raised. Plane xanthomata may present in the gingiva and, in this location, are usually associated with hyperlipidemia.24 An exceptional case has been described in an infant presenting with normolipemic papular and nodular lesions progressing to plane xanthomata and resulting in spontaneous resolution.25 Diffuse plane normolipemic xanthomata with mucosal and conjunctival involvement and aortic valve xanthomatosis may occur exceptionally.26 Lesions have been reported that clinically resembled plane xanthomata in a patient with systemic lupus erythematosus but histologically showed degeneration of collagen bundles with secondary fat deposition.27
Intertriginous xanthomata seen in patients with raised low density lipoproteins and pathognomonic of homozygous familial hypercholesterolemia present as yellow papules and plaques, often with a cobblestone appearance. These occur in the finger webspaces and to a lesser extent in the axillae and antecubital and popliteal fossae.2,28 They have a particularly high association with early and severe atherosclerosis. Intertriginous xanthomata may also rarely be seen in heterozygous familial hypercholesterolemia.29
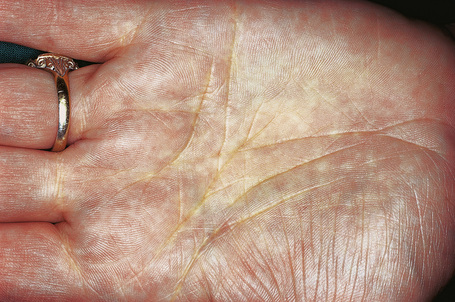
Planar xanthomata presenting as yellow–orange macules in the skin creases of the palm and fingers (xanthoma striatum palmare) are diagnostic of familial dysbetalipoproteinemia (HLP type III, broad beta disease) (Fig. 13.20), which is due to an abnormality of the apoprotein ApoE (homozygous ApoE2/E2).1,2,30 This results in impaired uptake of lipoprotein remnant particles by the liver and macrophages with resultant hyperlipoproteinemia and increased atherogenesis.29 Interestingly, the tendency to familial dysbetalipoproteinemia is present in 1% of the population, but a second lipid abnormality appears to be necessary to induce symptoms.29
Plane xanthomata of cholestasis, for example due to primary biliary cirrhosis and biliary atresia, present as well-demarcated, beige–orange plaques that are particularly found on the hands and feet, but may occur elsewhere.2 They can also develop in patients with diabetes mellitus and have been described in the setting of cholestasis resulting from chronic graft-versus-host disease.31
Planar xanthomata have also been described as a feature of HDL deficiency.32
Verruciform xanthoma
Clinical features
The verruciform xanthoma is an uncommon, asymptomatic lesion, which occurs predominantly in the oral cavity of adults in their fifth or sixth decade and shows a male predilection (1.7:1).1–5 It is most often found on the premolar gingiva of the mandible or maxilla.1 At this site it usually produces a solitary, well-circumscribed, asymptomatic, erythematous or yellow–tan lesion, 3–20 mm in diameter, which may be papillomatous or ulcerated. The patients are normolipidemic. The clinical differential diagnosis includes viral warts, leukoplakia, and squamous cell carcinoma.
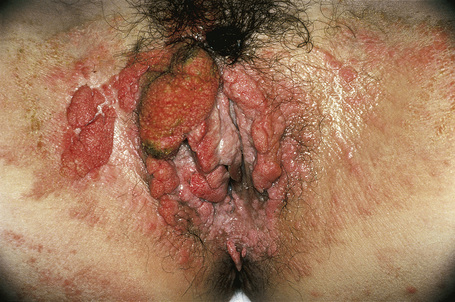
Verruciform xanthomata of the skin, which are extremely rare, have been described at a variety of sites including the ear, nose, and digits.6–9 Most cases described, however, have arisen on anogenital skin (Fig. 13.24).10–16 It may also develop as a reactive phenomenon within epidermolytic acanthoma, seborrheic keratoses, and epidermal nevi (including patients with inflammatory linear verrucous epidermal nevus or with the epidermal nevus syndrome) and has been recorded as a complication of lymphedema.9,17–22 It has been described in association with longstanding discoid lupus erythematosus, complicating ulceration in epidermolysis bullosa, and in association with squamous cell carcinoma of the penis.23–27 Occasionally, verruciform xanthomata are multifocal.6 Such a case has been described as multiple lesions in the upper aerodigestive tract of a child with a systemic lipid storage disease.28 Multiple verruciform xanthomata have also been reported in the anogenital region several years following necrotizing fasciitis of the perineum and in the oral mucosa in a patient with chronic graft-versus-host disease.29,30 A rare case of disseminated lesions has been described on the hands, feet, and anogenital region.31
Pathogenesis and histological features
The etiology and pathogenesis of the verruciform xanthoma are unknown. Originally, a viral infection by human papillomavirus (HPV) was suspected. Although most studies have not demonstrated definitive evidence to support this hypothesis, there are isolated reports demonstrating HPV DNA by polymerase chain reaction (PCR) in the lesions.32–35 Additionally, it has been suggested that keratinocyte necrosis may lead to the release of intracellular lipids, with resultant macrophage influx and xanthomatization.3,34,36 The inciting event leading to keratinocyte necrosis has not been identified. Immunohistochemical and electron microscopic studies tend to give support to this latter hypothesis (see below).
Verruciform xanthoma is an exophytic lesion characterized by massive but regular epidermal proliferation, parakeratosis, and hyperkeratosis (Fig. 13.25).32 Neutrophils, neutrophilic debris, and bacterial colonies may be evident in the parakeratotic stratum corneum. The acanthosis is associated with uniform, bulbous epidermal ridges, all of which penetrate to the same depth, giving a characteristically level lower border. The expanded ridges are associated with marked central keratinocyte necrosis and a heavy neutrophil polymorph inflammatory cell infiltrate (Fig. 13.26). There is no epithelial atypia and viral inclusions are invariably absent. The accentuated papillary dermis between the elongated epidermal ridges contains large numbers of eosinophilic foamy to granular xanthoma cells, which stain positively with lipid stains, but not usually with the diastase–periodic acid-Schiff (PAS) technique (Fig. 13.27). No foreign body or Touton giant cells are present. At the base of the lesion the epidermis may show focal basal cell hydropic degeneration associated with patchy loss of basement membrane. The reticular dermis deep to the lesion often contains a moderately dense lymphocyte–plasma cell infiltrate, which at the edge of the lesion sometimes adopts a lichenoid distribution. Typically,vascular ectasia is seen beneath the lesion.
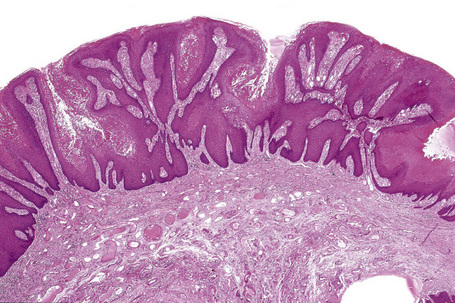
Fig. 13.25 Verruciform xanthoma: there is marked acanthosis, hyperkeratosis, and a level lower border.
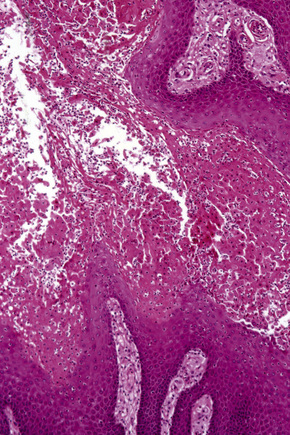
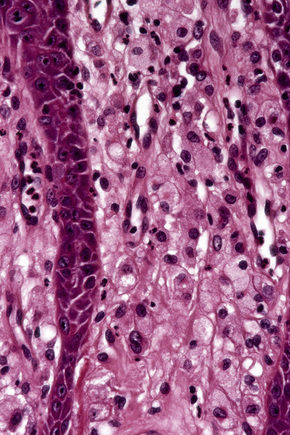
Fig. 13.27 Verruciform xanthoma: in the papillary dermis there is an infiltrate of uniform xanthoma cells.
By immunohistochemistry, fully formed foamy cells are negative for histiocytic markers including factor XIIIa, Mac 387, Ham-56, and KP1.34,37 Cells with incompletely lipidized cytoplasm show diffuse positivity for KP1 and weak positivity for FXIIIa and keratin. Cells with little cytoplasmic lipid are diffusely positive for FXIIIa and weakly positive for keratin. Nonlipidized cells located in the periphery of the infiltrate are diffusely positive for FXIIIa only. This staining pattern has led to the suggestion that FXIIIa-positive dermal dendritic cells play an active role in the formation of the lipid cells seen in this condition.34 It also tends to give further support to the role of damaged keratinocytes in the pathogenesis of verruciform xanthoma.34 The mechanism of keratinocyte damage is not fully elucidated. One theory proposes that the macrophages play an active role in keratinocyte cleavage and keratinolysis with secondary release of epithelial lipid. Macrophage recruitment is postulated to occur as a consequence of CD8-positive T cells present in the submucosa.38,39
Ultrastructural studies have revealed histiocytes containing numerous nonmembrane-bound lipid droplets, lysosomes, and myelin figures.11,40 Smaller numbers of these lipid inclusions may be found in the overlying keratinocytes and in the intercellular space. In one report, basal melanocytes were found to contain conspicuous lipid droplets.41 This was accompanied by evidence that the latter had been released into the basal intercellular space in association with disruption of the basal lamina, thereby providing a source for the lipid within the dermal macrophages.
Differential diagnosis
Angiokeratoma corporis diffusum
Clinical features
Angiokeratoma corporis diffusum (Anderson-Fabry’s disease) is a sex-linked recessive disorder of glycosphingolipid metabolism with a high mortality. It is very rare with an approximate incidence of 1 in 200 000.1 Deficiency of the lysosomal enzyme α-galactosidase A leads to the widespread accumulation of neutral glycolipids, mainly globotriaosylceramide (GB3, ceramidetrihexoside), and elevated urinary trihexoxylceramide levels.1–6
Globotriaosylceramide is normally broken down by α-galactosidase A to produce galactose and lactosylceramide. The full-blown syndrome is normally seen only in men, since female carriers have 15–40% greater enzyme activity than their male siblings or offspring. Heterozygotes, however, usually display abnormal ophthalmological and ultrastructural features.7 Occasionally, heterozygous females may manifest signs and symptoms due to extreme X inactivation (lyonization) of the healthy X chromosome.7,8 Cutaneous lesions are believed to occur in about 20% of heterozygous females.9
In males, the disease normally presents in childhood as episodes of excruciating intermittent pain, frequently in the fingers and toes.10 The attacks may be accompanied by fever, edema, and malaise. Patients may also have hypohidrosis, acroparesthesiae, and peripheral vasomotor disturbance affecting the heart, kidney, and central nervous system. Heat intolerance and telangiectases of the ears are often present early in the course of the disease.11
The characteristic angiokeratomata develop after puberty and present as tiny red–black bilaterally symmetrical papules, 0.5–2 mm in diameter, with slight hyperkeratosis.10 Lesions are typically seen in the bathing trunk distribution including the thighs, buttocks, lower back, penis, and scrotum, although occasional lesions may also be seen on the trunk or buccal mucosa (Figs. 13.28, 13.29). The number of angiokeratomata is highly variable. Atypical cases can present with an oligosymptomatic phenotype which includes only very few cutaneous angiokeratomata and asymptomatic involvement of organs such as the kidney and the heart.12 A female heterozygote with multiple nonkeratotic cutaneous angiomas has also been described.13 Nonvascular proliferations have been reported in patients with Fabry’s disease, including polyarteritis nodosa and leg ulcers.14,15 Telangiectasias develop in up to one-fourth of affected males.16,17 A recent study correlated the presence of angiokeratomas and telangiectasias with disease severity.16
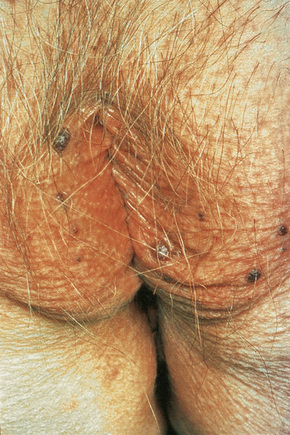
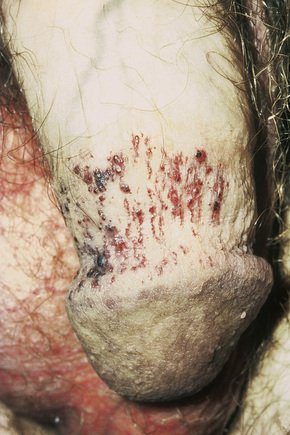
Fig. 13.29 Angiokeratoma corporis diffusum: conspicuous angiokeratomata on the penis, a commonly affected site.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
In a patient in whom the diagnosis is suspected, confirmation can usually be obtained by an ophthalmic examination. The conjunctival vessels may be tortuous or aneurysmal, as may the retinal vessels, and slit-lamp examination of the eyes reveals characteristic whorled, corneal linear opacities (verticillate cornea) (Fig. 13.30). Enzyme assay of α-galactosidase A can be performed using peripheral leukocytes or cutaneous fibroblasts. Hair root analysis has been recommended for the detection of heterozygotes.18
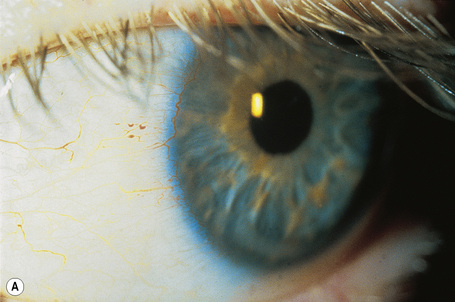
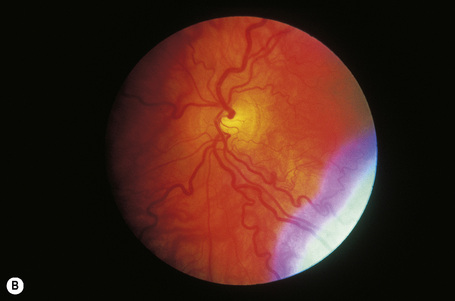
Fig. 13.30 Angiokeratoma corporis diffusum: (A) tortuous conjunctival vessels; (B) tortuous retinal vessels.
By courtesy of S. Parker, MD, St Thomas’ Hospital, London, UK.
Affected males can develop transient cerebrovascular accidents, but one of the most common causes of death is renal failure.19,20 In the early stages, proteinuria is seen and microscopy of the urinary sediment sometimes reveals characteristic lipid-laden cells even before proteinuria develops (Fig. 13.31). Electron microscopy may reveal the typical inclusions (Fig. 13.32).
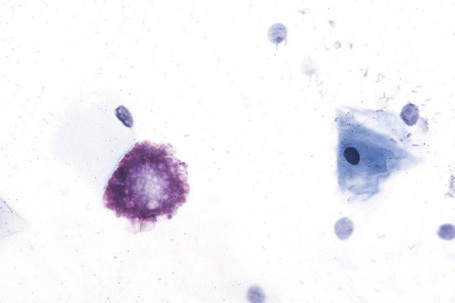
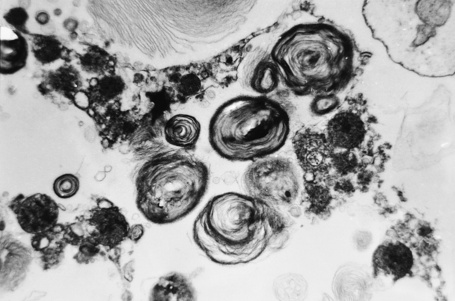
Cardiac involvement is found in approximately 20% of patients.21 Glycosphingolipid deposits in the conducting system, myocardium, endocardium, and valves may give rise to angina, electrocardiographic abnormalities, hypertrophic cardiomyopathy, hypertension, mitral valve incompetence, and aortic medial degeneration.22–25 Cardiovascular disease was found to be the most common cause of death in patients in a recent study of the Fabry registry.26
Oral and dental abnormalities are more common than previously realized and include the presence of cysts/pseudocysts of the maxillary sinuses and maxillary prognathism.27
Pathogenesis and histological features
A variety of genetic defects has been identified including point mutations, gene rearrangements, and deletions.28–31 A symptomatic heterozygous female Fabry’s disease patient without detectable mutation in the α-galactosidase gene has recently been described.32
The skin lesions are composed of ectatic blood-filled vessels in the papillary dermis, associated with slight hyperkeratosis (Figs. 13.33, 13.34). A characteristic feature is vacuolation of endothelial cells due to lipid deposits. The latter are doubly refractile and can usually be demonstrated in frozen material tissue sections. They may also be identified in toluidine blue-stained material. On electron microscopy, lamellar electron-dense inclusion bodies are present within endothelial cells, pericytes, smooth muscle cells, fibroblasts, sweat gland epithelium, and macrophages. It is believed that these are due to lipid deposition within lysosomes (Fig. 13.35). Lamellar bodies have also been identified in the endothelial cells of affected vessels with polyarteritis nodosa in a patient with Fabry’s disease.14
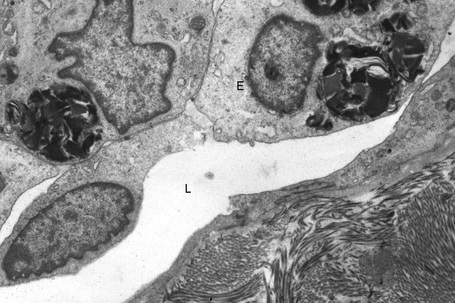
Differential diagnosis
Other forms of angiokeratomata, for example those of Mibelli or Fordyce, should be clinically distinguishable by their site and distribution although their histopathological appearances are identical. It should be noted, however, that diffuse angiokeratomata may also be seen in fucosidosis, α-galactosidosis, sialidosis, aspartylglycosaminuria, α-N- acetylgalactosaminidase deficiency (Kanzaki disease), human beta-mannosidosis, adult-onset GM1 gangliosidosis, and indeed, diffuse angiokeratomata of a benign type may occur in patients with normal enzyme activities.33–38 Widespread angiokeratomata have also been described as an exceptional finding in tuberous sclerosis.39
The amyloidoses
Amyloidosis is characterized by the extracellular deposition of a protein associated with particular tinctorial and ultrastructural properties. The amyloidoses are classified according to whether the amyloid deposition is systemic or localized (Table 13.3).
Table 13.3 Classification of the amyloidoses
| Systemic amyloidosis | Localized amyloidosis |
|---|---|
| Primary (due to an occult plasma cell dyscrasia) | Organs other than the skin* |
| Myeloma associated | Primary cutaneous |
| Secondary | Lichen, macular and biphasic |
| Hemodialysis associated | Secondary cutaneous |
| Heredofamilial† | Associated with neoplasms, porokeratosis and PUVA therapy |
| Amyloid elastosis | Familial cutaneous Nodular |
* Not discussed further in this chapter.
† Including familial Mediterranean fever, Muckle–Wells syndrome, familial amyloidotic polyneuropathy.
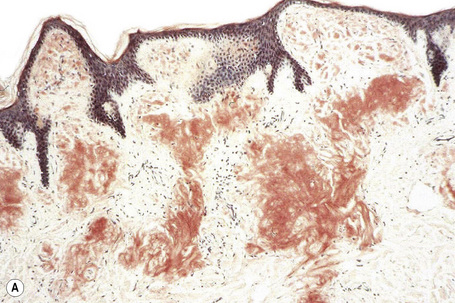
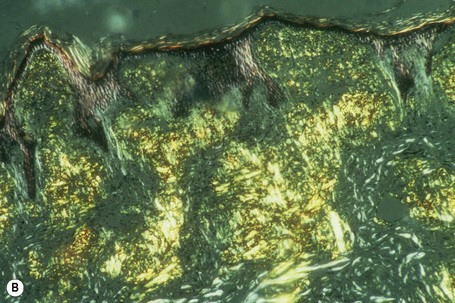
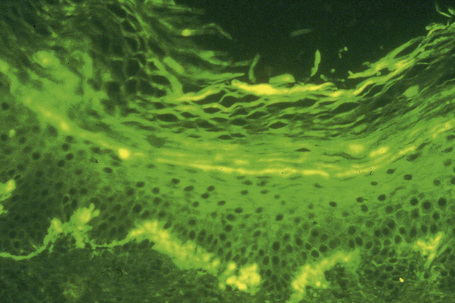
The most characteristic staining patterns of amyloid are seen with Congo red or Dylon (cotton dye pagoda red No. 9), which show apple-green birefringence under polarized light (Fig. 13.36).1 Unfortunately, this is not specific, and green birefringence may also be seen with collagen and in colloid milium, porphyria, and lipoid proteinosis. Amyloid deposits, which are PAS positive, may also be identified by the cotton dye Sirius red, or metachromatically using methyl or cresyl violet.2 Further confirmatory evidence can be obtained by staining with thioflavine-T and examination using fluorescence microscopy or by immunocytochemistry (see below) (Fig. 13.37).
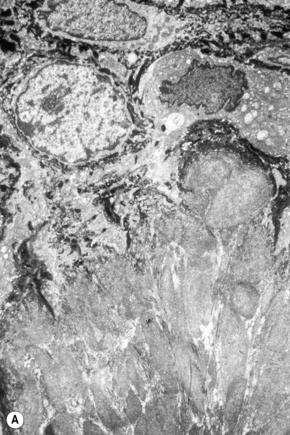
Amyloid shows characteristic and specific electron microscopic features of rigid, straight, nonbranching amyloid filaments with a diameter of 6–10 nm showing a hollow core on cross-section (Fig. 13.38).3 They are haphazardly distributed, lack the cross-banding of collagen, and are embedded in an electron-dense amorphous ground substance, which is probably composed of polysaccharides.
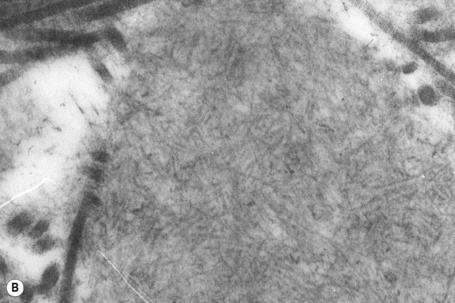
X-ray diffraction and infrared spectroscopy reveal a beta-pleated antiparallel configuration.4,5 Fibrils with a beta-pleated configuration are insoluble and highly resistant to proteolysis. This, combined with a lack of immunogenicity, results in their persistence at the site of deposition and subsequent tissue-damaging effects.
All forms of amyloid contain up to 14% by dry weight of a nonfibrillary protein, the serum amyloid P (SAP) component.2,6 The function of SAP is unknown, but it has been suggested that it may be primarily involved in the deposition and maintenance of the fibrillary components.2 Its presence, identified immunohistochemically, is a useful adjunct to the diagnosis of amyloidosis.7 However, it should be appreciated that the antibody also labels degenerate elastic fibers. The fibrillary component, however, may be derived in very different ways in each of the recognized types of amyloidosis: 8
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


