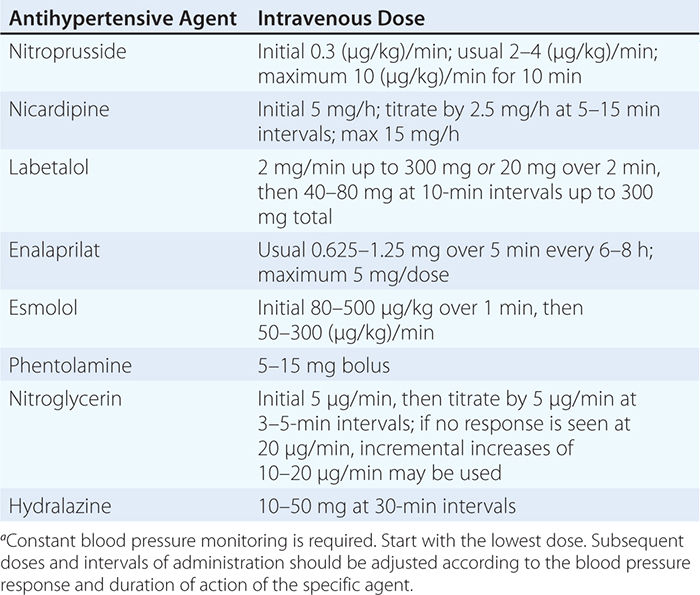
USUAL INTRAVENOUS DOSES OF ANTIHYPERTENSIVE AGENTS USED IN HYPERTENSIVE EMERGENCIESa |
Malignant hypertension is a syndrome associated with an abrupt increase of blood pressure in a patient with underlying hypertension or related to the sudden onset of hypertension in a previously normotensive individual. The absolute level of blood pressure is not as important as its rate of rise. Pathologically, the syndrome is associated with diffuse necrotizing vasculitis, arteriolar thrombi, and fibrin deposition in arteriolar walls. Fibrinoid necrosis has been observed in arterioles of kidney, brain, retina, and other organs. Clinically, the syndrome is recognized by progressive retinopathy (arteriolar spasm, hemorrhages, exudates, and papilledema), deteriorating renal function with proteinuria, microangiopathic hemolytic anemia, and encephalopathy. Historic inquiry should include questions about the use of monoamine oxidase inhibitors and recreational drugs (e.g., cocaine, amphetamines).
Although blood pressure should be lowered rapidly in patients with hypertensive encephalopathy, there are inherent risks of overly aggressive therapy. In hypertensive individuals, the upper and lower limits of autoregulation of cerebral blood flow are shifted to higher levels of arterial pressure, and rapid lowering of blood pressure to below the lower limit of autoregulation may precipitate cerebral ischemia or infarction as a consequence of decreased cerebral blood flow. Renal and coronary blood flows also may decrease with overly aggressive acute therapy. The initial goal of therapy is to reduce mean arterial blood pressure by no more than 25% within minutes to 2 h or to a blood pressure in the range of 160/100–110 mmHg. This may be accomplished with IV nitroprusside, a short-acting vasodilator with a rapid onset of action that allows for minute-to-minute control of blood pressure. Parenteral labetalol and nicardipine are also effective agents for the treatment of hypertensive encephalopathy.
In patients with malignant hypertension without encephalopathy or another catastrophic event, it is preferable to reduce blood pressure over hours or longer rather than minutes. This goal may effectively be achieved initially with frequent dosing of short-acting oral agents such as captopril, clonidine, and labetalol.
Acute, transient blood pressure elevations that last days to weeks frequently occur after thrombotic and hemorrhagic strokes. Autoregulation of cerebral blood flow is impaired in ischemic cerebral tissue, and higher arterial pressures may be required to maintain cerebral blood flow. Although specific blood pressure targets have not been defined for patients with acute cerebrovascular events, aggressive reductions of blood pressure are to be avoided. With the increasing availability of improved methods for measuring cerebral blood flow (using CT technology), studies are in progress to evaluate the effects of different classes of antihypertensive agents on both blood pressure and cerebral blood flow after an acute stroke. Currently, in the absence of other indications for acute therapy, for patients with cerebral infarction who are not candidates for thrombolytic therapy, one recommended guideline is to institute antihypertensive therapy only for patients with a systolic blood pressure >220 mmHg or a diastolic blood pressure >130 mmHg. If thrombolytic therapy is to be used, the recommended goal blood pressure is <185 mmHg systolic pressure and <110 mmHg diastolic pressure. In patients with hemorrhagic stroke, suggested guidelines for initiating antihypertensive therapy are systolic >180 mmHg or diastolic pressure >130 mmHg. The management of hypertension after subarachnoid hemorrhage is controversial. Cautious reduction of blood pressure is indicated if mean arterial pressure is >130 mmHg.
In addition to pheochromocytoma, an adrenergic crisis due to catecholamine excess may be related to cocaine or amphetamine overdose, clonidine withdrawal, acute spinal cord injuries, and an interaction of tyramine-containing compounds with monoamine oxidase inhibitors. These patients may be treated with phentolamine or nitroprusside.
Treatment of hypertension in patients with acute aortic dissection is discussed in Chap. 301, and treatment of hypertension in pregnancy is discussed in Chap. 8.
299 | Renovascular Disease |
The renal vasculature is unusually complex with rich arteriolar flow to the cortex in excess of metabolic requirements, consistent with its primary function as a filtering organ. After delivering blood to cortical glomeruli, the postglomerular circulation supplies deeper medullary segments that support energy-dependent solute transport at multiple levels of the renal tubule. These postglomerular vessels carry less blood, and high oxygen consumption leaves the deeper medullary regions at the margin of hypoxemia. Vascular disorders that commonly threaten the blood supply of the kidney include large-vessel atherosclerosis, fibromuscular diseases, and embolic disorders. Microvascular injury, including inflammatory and primary hematologic disorders, is described in Chap. 341.
The glomerular capillary endothelium shares susceptibility to oxidative stress, pressure injury, and inflammation with other vascular territories. Rates of urinary albumin excretion (UAE) are predictive of systemic atherosclerotic disease events. Increased UAE may develop years before cardiovascular events. UAE and the risk of cardiovascular events are both reduced with pharmacologic therapy such as statins. Experimental studies demonstrate functional changes and rarefaction of renal microvessels under conditions of accelerated atherosclerosis and/or compromise of proximal perfusion pressures with large-vessel disease (Fig. 299-1).
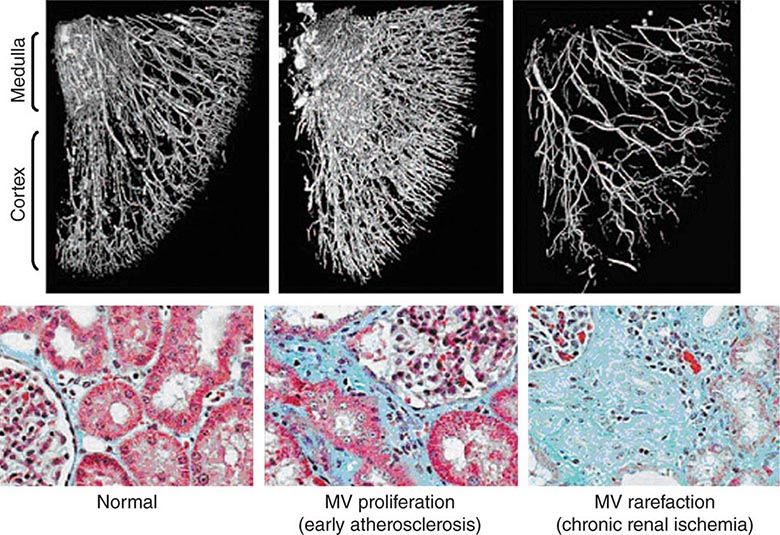
FIGURE 299-1 Examples of micro-CT images from vessels defined by radiopaque casts injected into the renal vasculature. These illustrate the complex, dense cortical capillary network supplying the kidney cortex that can either proliferate or succumb to rarefaction under the influence of atherosclerosis and/or occlusive disease. Changes in blood supply are followed by tubulointerstitial fibrosis and loss of kidney function. MV, microvascular. (From LO Lerman, AR Chade: Curr Opin Nephrol Hyper 18:160, 2009, with permission.)
MACROVASCULAR DISEASE
Large-vessel renal artery occlusive disease can result from extrinsic compression of the vessel, fibromuscular dysplasia, or, most commonly, atherosclerotic disease. Any disorder that reduces perfusion pressure to the kidney can activate mechanisms that tend to restore renal pressures at the expense of developing systemic hypertension. Because restoration of perfusion pressures can reverse these pathways, renal artery stenosis is considered a specifically treatable “secondary” cause of hypertension.
Renal artery stenosis is common and often has only minor hemodynamic effects. Fibromuscular dysplasia (FMD) is reported in 3–5% of normal subjects presenting as potential kidney donors without hypertension. It may present clinically with hypertension in younger individuals (between age 15 and 50), most often women. FMD does not often threaten kidney function, but sometimes produces total occlusion and can be associated with renal artery aneurysms. Atherosclerotic renal artery stenosis (ARAS) is common in the general population (6.8% of a community-based sample above age 65), and the prevalence increases with age and for patients with other vascular conditions such as coronary artery disease (18–23%) and/or peripheral aortic or lower extremity disease (>30%). If untreated, ARAS progresses in nearly 50% of cases over a 5-year period, sometimes to total occlusion. Intensive treatment of arterial blood pressure and statin therapy appear to slow these rates and improve clinical outcomes.
Critical levels of stenosis lead to a reduction in perfusion pressure that activates the renin-angiotensin system, reduces sodium excretion, and activates sympathetic adrenergic pathways. These events lead to systemic hypertension characterized by angiotensin dependence in the early stages, widely varying pressures, loss of circadian blood pressure (BP) rhythms, and accelerated target organ injury, including left ventricular hypertrophy and renal fibrosis. Renovascular hypertension can be treated with agents that block the renin-angiotensin system and other drugs that modify these pressor pathways. It can also be treated with restoration of renal blood flow by either endovascular or surgical revascularization. Most patients require continued antihypertensive drug therapy because revascularization alone rarely lowers BP to normal.
ARAS and systemic hypertension tend to affect both the post-stenotic and contralateral kidneys, reducing overall glomerular filtration rate (GFR) in ARAS. When kidney function is threatened by large-vessel disease primarily, it has been labeled ischemic nephropathy. Moderately reduced blood flow that develops gradually is associated with reduced GFR and limited oxygen consumption with preserved tissue oxygenation. Hence, kidney function can remain stable during medical therapy, sometimes for years. With more advanced disease, reductions in cortical perfusion and frank tissue hypoxia develop. Unlike FMD, ARAS develops in patients with other risk factors for atherosclerosis and is commonly superimposed upon preexisting small-vessel disease in the kidney resulting from hypertension, aging, and diabetes. Nearly 85% of patients considered for renal revascularization have stage 3–5 chronic kidney disease (CKD) with GFR below 60 mL/min per 1.73 m2. The presence of ARAS is a strong predictor of morbidity- and mortality-related cardiovascular events, independent of whether renal revascularization is undertaken.
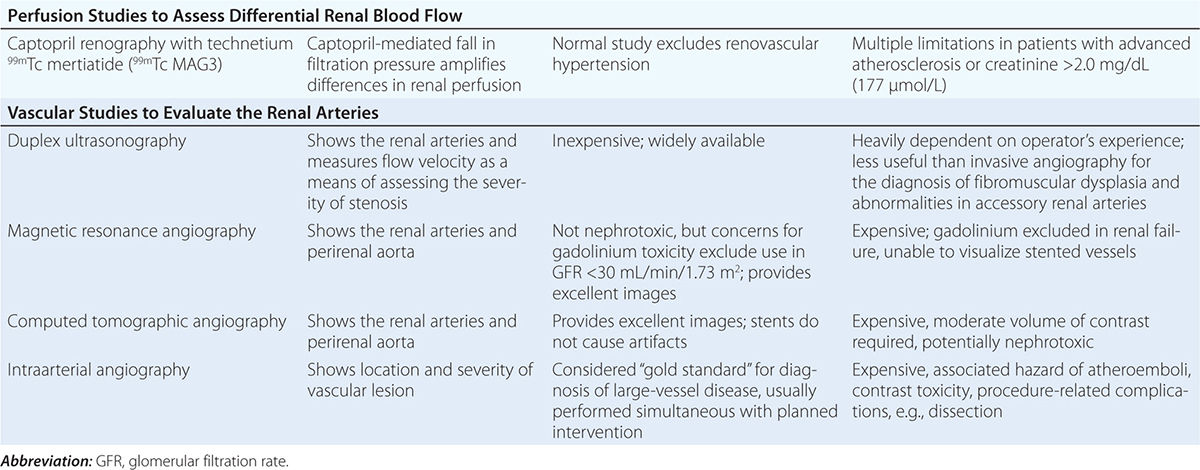
Diagnostic approaches to renal artery stenosis depend partly on the specific issues to be addressed. Noninvasive characterization of the renal vasculature may be achieved by several techniques, summarized in Table 299-1. Although activation of the renin-angiotensin system is a key step in developing renovascular hypertension, it is transient. Levels of renin activity are therefore subject to timing, the effects of drugs, and sodium intake, and do not reliably predict the response to vascular therapy. Renal artery velocities by Doppler ultrasound above 200 cm/s generally predict hemodynamically important lesions (above 60% vessel lumen occlusion), although treatment trials require velocity above 300 cm/s to avoid false positives. The renal resistive index has predictive value regarding the viability of the kidney. It remains operator- and institution-dependent, however. Captopril-enhanced renography has a strong negative predictive value when entirely normal. Magnetic resonance angiography (MRA) is now less often used, as gadolinium contrast has been associated with nephrogenic systemic fibrosis. Contrast-enhanced computed tomography (CT) with vascular reconstruction provides excellent vascular images and functional assessment, but carries a small risk of contrast toxicity.
SUMMARY OF IMAGING MODALITIES FOR EVALUATING THE KIDNEY VASCULATURE |
TREATMENT RENAL ARTERY STENOSIS
While restoring renal blood flow and perfusion seems intuitively beneficial for high-grade occlusive lesions, revascularization procedures also pose hazards and expense. Patients with FMD are commonly younger females with otherwise normal vessels and a long life expectancy. These patients often respond well to percutaneous renal artery angioplasty. If BP can be controlled to goal levels and kidney function remains stable in patients with ARAS, it may be argued that medical therapy with follow-up for disease progression is equally effective. Prospective trials up to now have failed to identify compelling benefits for interventional procedures regarding short-term results of BP and renal function, and long-term studies regarding cardiovascular outcomes, such as stroke, congestive heart failure, myocardial infarction, and end-stage renal failure, are not yet complete. Medical therapy should include blockade of the renin-angiotensin system, attainment of goal BPs, cessation of tobacco, statins, and aspirin. Renal revascularization is now often reserved for patients failing medical therapy or developing additional complications.
Techniques of renal revascularization are improving. With experienced operators, major complications occur in about 9% of cases, including renal artery dissection, capsular perforation, hemorrhage, and occasional atheroembolic disease. Although not common, atheroembolic disease can be catastrophic and accelerate both hypertension and kidney failure, precisely the events that revascularization is intended to prevent. Although renal blood flow usually can be restored by endovascular stenting, recovery of renal function is limited to about 25% of cases, with no change in 50% and some deterioration evident in others. Patients with rapid loss of kidney function, sometimes associated with antihypertensive drug therapy, or with vascular disease affecting the entire functioning kidney mass are more likely to recover function after restoring blood flow. When hypertension is refractory to effective therapy, revascularization offers real benefits. Table 299-2 summarizes currently accepted guidelines for considering renal revascularization.
CLINICAL FACTORS FAVORING MEDICAL THERAPY AND REVASCULARIZATION OR SURVEILLANCE FOR RENAL ARTERY STENOSIS |
Abbreviations: ACE, angiotensin-converting enzyme; ARBs, angiotensin receptor blockers; GFR, glomerular filtration rate.
ATHEROEMBOLIC RENAL DISEASE
Emboli to the kidneys arise most frequently as a result of cholesterol crystals breaking free of atherosclerotic vascular plaque and lodging in downstream microvessels. Most clinical atheroembolic events follow angiographic procedures, often of the coronary vessels. It has been argued that nearly all vascular interventional procedures lead to plaque fracture and release of microemboli, but clinical manifestations develop only in a fraction of these. The incidence of clinical atheroemboli has been increasing with more vascular procedures and longer life spans. Atheroembolic renal disease is suspected in more than 3% of elderly subjects with end-stage renal disease (ESRD) and is likely underdiagnosed. It is more frequent in males with a history of diabetes, hypertension, and ischemic cardiac disease. Atheroemboli in the kidney are strongly associated with aortic aneurysmal disease and renal artery stenosis. Most clinical cases can be linked to precipitating events, such as angiography, vascular surgery, anticoagulation with heparin, thrombolytic therapy, or trauma. Clinical manifestations of this syndrome commonly develop between 1 and 14 days after an inciting event and may continue to develop for weeks thereafter. Systemic embolic disease manifestations, such as fever, abdominal pain, and weight loss, are present in less than half of patients, although cutaneous manifestations including livedo reticularis and localized toe gangrene may be more common. Worsening hypertension and deteriorating kidney function are common, sometimes reaching a malignant phase. Progressive renal failure can occur and require dialytic support. These cases often develop after a stuttering onset over many weeks and have an ominous prognosis. Mortality rate after 1 year reaches 38%, and although some may eventually recover sufficiently to no longer require dialysis, many do not.
Beyond the clinical manifestations above, laboratory findings include rising creatinine, transient eosinophilia (60–80%), elevated sedimentation rate, and hypocomplementemia (15%). Establishing this diagnosis can be difficult and is often by exclusion. Definitive diagnosis depends on kidney biopsy demonstrating microvessel occlusion with cholesterol crystals that leave a “cleft” in the vessel. Biopsies obtained from patients undergoing surgical revascularization of the kidney indicate that silent cholesterol emboli are frequently present before any further manipulation is performed.
No effective therapy is available for atheroembolic disease once it has developed. Withdrawal of anticoagulation is recommended. Late recovery of kidney function after supportive measures sometimes occurs, and statin therapy may improve outcome. The role of embolic protection devices in the renal circulation is unclear, but a few prospective trials have failed to demonstrate major benefits. These devices are limited to distal protection during the endovascular procedure and offer no protection from embolic debris after removal.
THROMBOEMBOLIC RENAL DISEASE
Thrombotic occlusion of renal vessels or branch arteries can lead to declining renal function and hypertension. It is difficult to diagnose and is often overlooked, especially in elderly patients. Thrombosis can develop as a result of local vessel abnormalities, such as local dissection, trauma, or inflammatory vasculitis. Local microdissections sometimes lead to patchy, transient areas of infarctions labeled “segmental arteriolar mediolysis.” Although hypercoagulability conditions sometimes present as renal artery thrombosis, this is rare. It can also derive from distant embolic events, e.g., the left atrium in patients with atrial fibrillation or from fat emboli originating from traumatized tissue, most commonly large bone fractures. Cardiac sources include vegetations from subacute bacterial endocarditis. Systemic emboli to the kidneys may also arise from the venous circulation if right-to-left shunting occurs, e.g., through a patent foramen ovale.
Clinical manifestations vary depending on the rapidity of onset and extent of occlusion. Acute arterial thrombosis may produce flank pain, fever, leukocytosis, nausea, and vomiting. If kidney infarction results, enzymes such as lactate dehydrogenase (LDH) rise to extreme levels. If both kidneys are affected, renal function will decline precipitously with a drop in urine output. If a single kidney is involved, renal functional changes may be minor. Hypertension related to sudden release of renin from ischemic tissue can develop rapidly, as long as some viable tissue in the “peri-infarct” border zone remains. If the infarct zone demarcates precisely, the rise in BP and renin activity may resolve. Diagnosis of renal infarction may be established by vascular imaging with MRI, CT angiography, or arteriography (Fig. 299-2).
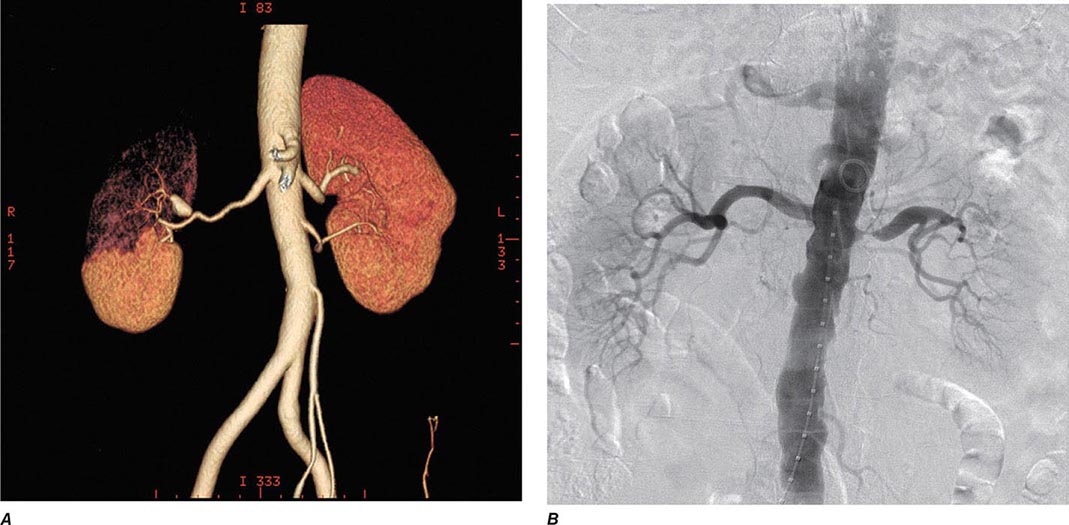
FIGURE 299-2 A. CT angiogram illustrating loss of circulation to the upper pole of the right kidney in a patient with fibromuscular disease and a renal artery aneurysm. Activation of the renin-angiotensin system produced rapidly developing hypertension. B. Angiogram illustrating high-grade renal artery stenosis affecting the left kidney. This lesion is often part of widespread atherosclerosis and sometimes is an extension of aortic plaque. This lesion develops in older individuals with preexisting atherosclerotic risk factors.
MANAGEMENT OF ARTERIAL THROMBOSIS OF THE KIDNEY
Options for interventions of newly detected arterial occlusion include surgical reconstruction, anticoagulation, thrombolytic therapy, endovascular procedures, and supportive care, particularly antihypertensive drug therapy. Application of these methods depends on the patient’s overall condition, the precipitating factors (e.g., local trauma or systemic illness), the magnitude of renal tissue and function at risk, and the likelihood of recurrent events in the future. For unilateral disease, e.g., arterial dissection with thrombosis, supportive care with anticoagulation may suffice. Acute, bilateral occlusion is potentially catastrophic, producing anuric renal failure. Depending on the precipitating event, surgical or thrombolytic therapies can sometimes restore kidney viability.
MICROVASCULAR INJURY ASSOCIATED WITH HYPERTENSION
ARTERIOLONEPHROSCLEROSIS
“Malignant” Hypertension Although BP rises with age, it has long been recognized that some individuals develop rapidly progressive BP elevations with target organ injury including retinal hemorrhages, encephalopathy, and declining kidney function. Placebo arms during the controlled trials of hypertension therapy identified progression to severe levels in 20% of subjects over 5 years. If untreated, patients with target organ injury including papilledema and declining kidney function suffered mortality rates in excess of 50% over 6–12 months, hence the designation “malignant.” Postmortem studies of such patients identified vascular lesions, designated “fibrinoid necrosis,” with breakdown of the vessel wall, deposition of eosinophilic material including fibrin, and a perivascular cellular infiltrate. A separate lesion was identified in the larger interlobular arteries in many patients with hyperplastic proliferation of the vascular wall cellular elements, deposition of collagen, and separation of layers, designated the “onionskin” lesion. For many of these patients, fibrinoid necrosis led to obliteration of glomeruli and loss of tubular structures. Progressive kidney failure ensued and, without dialysis support, led to early mortality in untreated malignant-phase hypertension. These vascular changes could develop with pressure-related injury from a variety of hypertensive pathways, including but not limited to activation of the renin-angiotensin system and severe vasospasm associated with catecholamine release. Occasionally, endothelial injury is sufficient to induce microangiopathic hemolysis, as discussed below.
Antihypertensive therapy is the mainstay of therapy for malignant hypertension. With effective BP reduction, manifestations of vascular injury including microangiopathic hemolysis and renal dysfunction can improve over time. Whereas series reported before the era of drug therapy suggested that 1-year mortality rates exceeded 90%, current survival over 5 years exceeds 50%.
Malignant hypertension is less common in Western countries, although it persists in parts of the world where medical care and antihypertensive drug therapy are less available. It most commonly develops in patients with treated hypertension who neglect to take medications or who may use vasospastic drugs, such as cocaine. Renal abnormalities typically include rising serum creatinine and occasionally hematuria and proteinuria. Biochemical findings may include evidence of hemolysis (anemia, schistocytes, and reticulocytosis) and changes associated with kidney failure. African-American males are more likely to develop rapidly progressive hypertension and kidney failure than are whites in the United States. Genetic polymorphisms (first identified as MYH9, but now thought to be APOL1) that are common in the African-American population predispose to subtle focal sclerosing glomerular disease, with severe hypertension developing at younger ages secondary to renal disease in this instance.
“Hypertensive Nephrosclerosis” Based on experience with malignant hypertension and epidemiologic evidence linking BP with long-term risks of kidney failure, it has long been assumed that lesser degrees of hypertension induce less severe, but prevalent, changes in kidney vessels and loss of kidney function. As a result, a large portion of patients reaching ESRD without a specific etiologic diagnosis are assigned the designation “hypertensive nephrosclerosis.” Pathologic examination commonly identifies afferent arteriolar thickening with deposition of homogeneous eosinophilic material (hyaline arteriolosclerosis) associated with narrowing of vascular lumina. Clinical manifestations include retinal vessel changes associated with hypertension (arteriolar narrowing, crossing changes), left ventricular hypertrophy, and elevated BP. The role of these vascular changes in kidney function is unclear. Postmortem and biopsy samples from normotensive kidney donors demonstrate similar vessel changes associated with aging, dyslipidemia, and glucose intolerance. Although BP reduction does slow progression of proteinuric kidney diseases and is warranted to reduce the excessive cardiovascular risks associated with CKD, antihypertensive therapy does not alter the course of kidney dysfunction identified specifically as hypertensive nephrosclerosis.
300 | Deep Venous Thrombosis and Pulmonary Thromboembolism |
EPIDEMIOLOGY
Venous thromboembolism (VTE) encompasses deep venous thrombosis (DVT) and pulmonary embolism (PE) and causes cardiovascular death and disability. In the United States, the Surgeon General estimates there are 100,000 to 180,000 deaths annually from PE and has declared that PE is the most common preventable cause of death among hospitalized patients. Survivors may succumb to the disabilities of chronic thromboembolic pulmonary hypertension or postthrombotic syndrome. Chronic thromboembolic pulmonary hypertension causes breathlessness, especially with exertion. Postthrombotic syndrome (also known as chronic venous insufficiency) damages the venous valves of the leg and causes ankle or calf swelling and leg aching, especially after prolonged standing. In its most severe form, postthrombotic syndrome causes skin ulceration (Fig. 300-1).
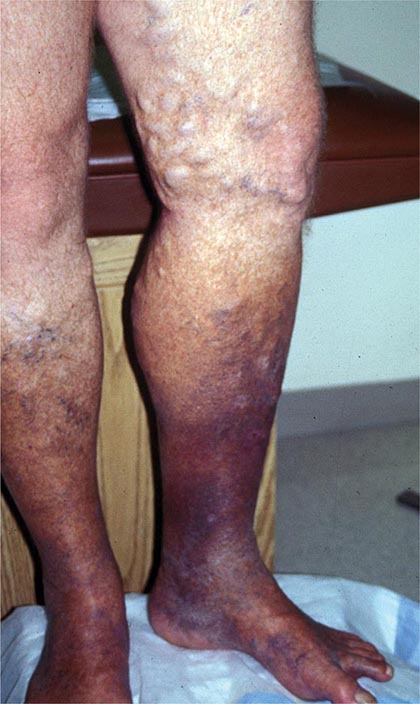
FIGURE 300-1 Skin ulceration in the lateral malleolus from postthrombotic syndrome of the leg.
PATHOPHYSIOLOGY
Inflammation and Platelet Activation Virchow’s triad of inflammation, hypercoagulability, and endothelial injury leads to recruitment of activated platelets, which release microparticles. These microparticles contain proinflammatory mediators that bind neutrophils, stimulating them to release their nuclear material and form web-like extracellular networks called neutrophil extracellular traps. These prothrombotic networks contain histones that stimulate platelet aggregation and promote platelet-dependent thrombin generation. Venous thrombi form and flourish in an environment of stasis, low oxygen tension, and upregulation of proinflammatory genes.
Prothrombotic States The two most common autosomal dominant genetic mutations are factor V Leiden, which causes resistance to the endogenous anticoagulant, activated protein C (which inactivates clotting factors V and VIII), and the prothrombin gene mutation, which increases the plasma prothrombin concentration (Chaps. 78 and 142). Antithrombin, protein C, and protein S are naturally occurring coagulation inhibitors. Deficiencies of these inhibitors are associated with VTE but are rare. Antiphospholipid antibody syndrome is the most common acquired cause of thrombophilia and is associated with venous or arterial thrombosis. Other common predisposing factors include cancer, obesity, cigarette smoking, systemic arterial hypertension, chronic obstructive pulmonary disease, chronic kidney disease, blood transfusion, long-haul air travel, air pollution, oral contraceptives, pregnancy, postmenopausal hormone replacement, surgery, and trauma.
Embolization When deep venous thrombi (Fig. 300-2) detach from their site of formation, they embolize to the vena cava, right atrium, and right ventricle, and lodge in the pulmonary arterial circulation, thereby causing acute PE. Paradoxically, these thrombi occasionally embolize to the arterial circulation through a patent foramen ovale or atrial septal defect. Many patients with PE have no evidence of DVT because the clot has already embolized to the lungs.
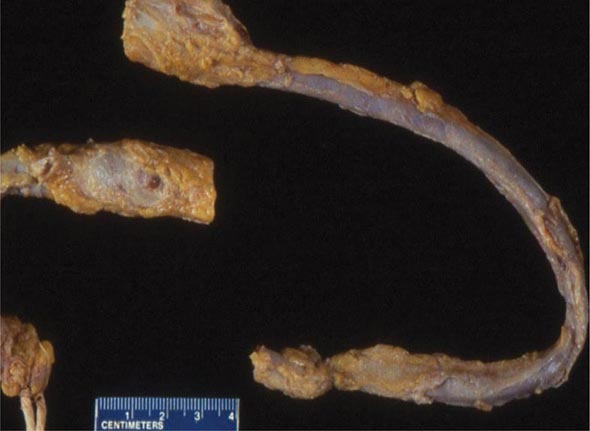
FIGURE 300-2 Deep venous thrombosis at autopsy.
Physiology The most common gas exchange abnormalities are arterial hypoxemia and an increased alveolar-arterial O2 tension gradient, which represents the inefficiency of O2 transfer across the lungs. Anatomic dead space increases because breathed gas does not enter gas exchange units of the lung. Physiologic dead space increases because ventilation to gas exchange units exceeds venous blood flow through the pulmonary capillaries.
Other pathophysiologic abnormalities include:
1. Increased pulmonary vascular resistance due to vascular obstruction or platelet secretion of vasoconstricting neurohumoral agents such as serotonin. Release of vasoactive mediators can produce ventilation-perfusion mismatching at sites remote from the embolus, thereby accounting for discordance between a small PE and a large alveolar-arterial O2 gradient.
2. Impaired gas exchange due to increased alveolar dead space from vascular obstruction, hypoxemia from alveolar hypoventilation relative to perfusion in the nonobstructed lung, right-to-left shunting, or impaired carbon monoxide transfer due to loss of gas exchange surface.
3. Alveolar hyperventilation due to reflex stimulation of irritant receptors.
4. Increased airway resistance due to constriction of airways distal to the bronchi.
5. Decreased pulmonary compliance due to lung edema, lung hemorrhage, or loss of surfactant.
Pulmonary Hypertension, Right Ventricular (RV) Dysfunction, and RV Microinfarction Pulmonary artery obstruction causes a rise in pulmonary artery pressure and in pulmonary vascular resistance. When RV wall tension rises, RV dilation and dysfunction ensue, with release of the cardiac biomarker, brain natriuretic peptide. The interventricular septum bulges into and compresses an intrinsically normal left ventricle (LV). Diastolic LV dysfunction reduces LV distensibility and impairs LV filling. Increased RV wall tension also compresses the right coronary artery, limits myocardial oxygen supply, and precipitates right coronary artery ischemia and RV microinfarction, with release of cardiac biomarkers such as troponin. Underfilling of the LV may lead to a fall in LV cardiac output and systemic arterial pressure, with consequent circulatory collapse and death.
CLASSIFICATION OF PULMONARY EMBOLISM AND DEEP VENOUS THROMBOSIS
Pulmonary Embolism Massive PE accounts for 5–10% of cases, and is characterized by extensive thrombosis affecting at least half of the pulmonary vasculature. Dyspnea, syncope, hypotension, and cyanosis are hallmarks of massive PE. Patients with massive PE may present in cardiogenic shock and can die from multisystem organ failure. Submassive PE accounts for 20–25% of patients, and is characterized by RV dysfunction despite normal systemic arterial pressure. The combination of right heart failure and release of cardiac biomarkers indicates an increased likelihood of clinical deterioration. Low-risk PE constitutes about 70–75% of cases. These patients have an excellent prognosis.
Deep Venous Thrombosis Lower extremity DVT usually begins in the calf and propagates proximally to the popliteal vein, femoral vein, and iliac veins. Leg DVT is about 10 times more common than upper extremity DVT, which is often precipitated by placement of pacemakers, internal cardiac defibrillators, or indwelling central venous catheters. The likelihood of upper extremity DVT increases as the catheter diameter and number of lumens increase. Superficial venous thrombosis usually presents with erythema, tenderness, and a “palpable cord.” Patients are at risk for extension of the thrombosis to the deep venous system.
DIAGNOSIS
Clinical Evaluation PE is known as “the Great Masquerader.” Diagnosis is difficult because symptoms and signs are nonspecific. The most common symptom is unexplained breathlessness. When occult PE occurs concomitantly with overt congestive heart failure or pneumonia, clinical improvement often fails to occur despite standard medical treatment of the concomitant illness. This scenario presents a clinical clue to the possible coexistence of PE.
With DVT, the most common symptom is a cramp or “charley horse” in the lower calf that persists and intensifies over several days. Point score criteria help estimate the clinical likelihood of DVT and PE (Table 300-1). Patients with a low-to-moderate likelihood of DVT or PE should undergo initial diagnostic evaluation with D-dimer testing alone (see “Blood Tests”) without obligatory imaging tests (Fig. 300-3). However, patients with a high clinical likelihood of VTE should skip D-dimer testing and undergo imaging as the next step in the diagnostic algorithm.
CLINICAL DECISION RULES |
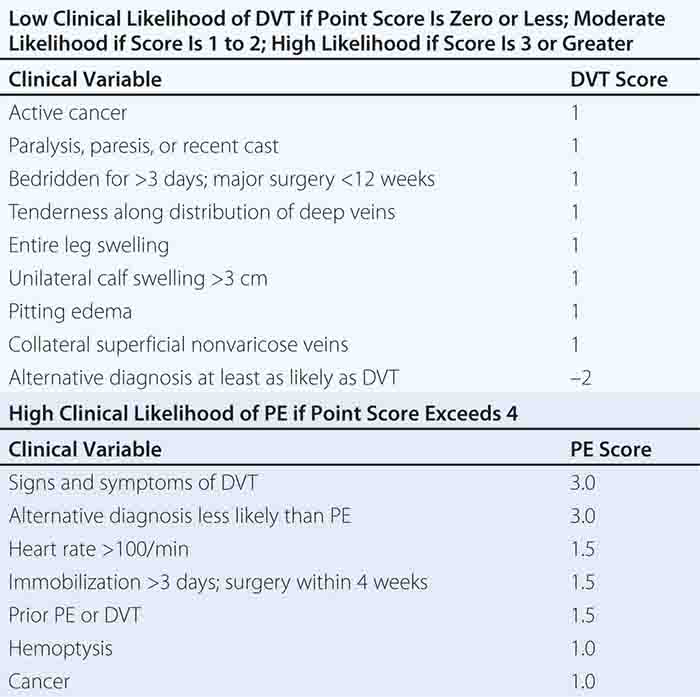
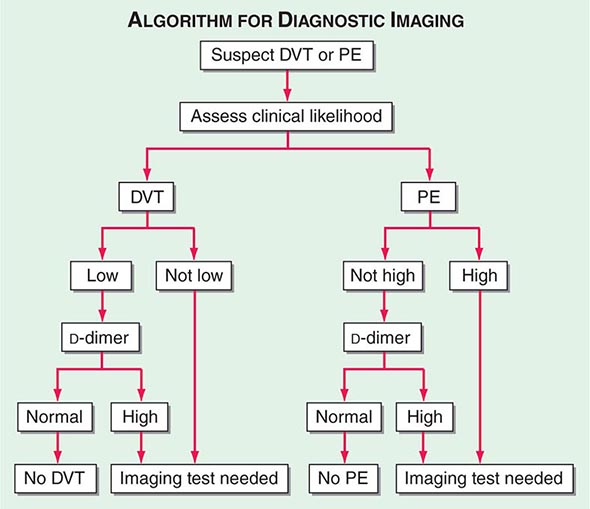
FIGURE 300-3 How to decide whether diagnostic imaging is needed. For assessment of clinical likelihood, see Table 300-1.
Clinical Pearls Not all leg pain is due to DVT, and not all dyspnea is due to PE (Table 300-2). Sudden, severe calf discomfort suggests a ruptured Baker’s cyst. Fever and chills usually herald cellulitis rather than DVT. Physical findings, if present, may consist only of mild palpation discomfort in the lower calf. However, massive DVT often presents with marked thigh swelling, tenderness, and erythema. If the leg is diffusely edematous, DVT is unlikely. More probable is an acute exacerbation of venous insufficiency due to postthrombotic syndrome. Upper extremity venous thrombosis may present with asymmetry in the supraclavicular fossa or in the circumference of the upper arms.
DIFFERENTIAL DIAGNOSIS |
Pulmonary infarction usually indicates a small PE. This condition is exquisitely painful because the thrombus lodges peripherally, near the innervation of pleural nerves. Nonthrombotic PE etiologies include fat embolism after pelvic or long bone fracture, tumor embolism, bone marrow, and air embolism. Cement embolism and bony fragment embolism can occur after total hip or knee replacement. Intravenous drug users may inject themselves with a wide array of substances that can embolize such as hair, talc, and cotton. Amniotic fluid embolism occurs when fetal membranes leak or tear at the placental margin.
Nonimaging Diagnostic Modalities • BLOOD TESTS The quantitative plasma D-dimer enzyme-linked immunosorbent assay (ELISA) rises in the presence of DVT or PE because of the breakdown of fibrin by plasmin. Elevation of D-dimer indicates endogenous although often clinically ineffective thrombolysis. The sensitivity of the D-dimer is >80% for DVT (including isolated calf DVT) and >95% for PE. The D-dimer is less sensitive for DVT than for PE because the DVT thrombus size is smaller. A normal D-dimer is a useful “rule out” test. However, the D-dimer assay is not specific. Levels increase in patients with myocardial infarction, pneumonia, sepsis, cancer, and the postoperative state and those in the second or third trimester of pregnancy. Therefore, D-dimer rarely has a useful role among hospitalized patients, because levels are frequently elevated due to systemic illness.
ELEVATED CARDIAC BIOMARKERS Serum troponin and plasma heart-type fatty acid–binding protein levels increase because of RV microinfarction. Myocardial stretch causes release of brain natriuretic peptide or NT-pro-brain natriuretic peptide.
ELECTROCARDIOGRAM The most frequently cited abnormality, in addition to sinus tachycardia, is the S1Q3T3 sign: an S wave in lead I, a Q wave in lead III, and an inverted T wave in lead III (Chap. 268). This finding is relatively specific but insensitive. RV strain and ischemia cause the most common abnormality, T-wave inversion in leads V1 to V4.
Noninvasive Imaging Modalities • VENOUS ULTRASONOGRAPHY Ultrasonography of the deep venous system relies on loss of vein compressibility as the primary criterion for DVT. When a normal vein is imaged in cross-section, it readily collapses with gentle manual pressure from the ultrasound transducer. This creates the illusion of a “wink.” With acute DVT, the vein loses its compressibility because of passive distention by acute thrombus. The diagnosis of acute DVT is even more secure when thrombus is directly visualized. It appears homogeneous and has low echogenicity (Fig. 300-4). The vein itself often appears mildly dilated, and collateral channels may be absent.
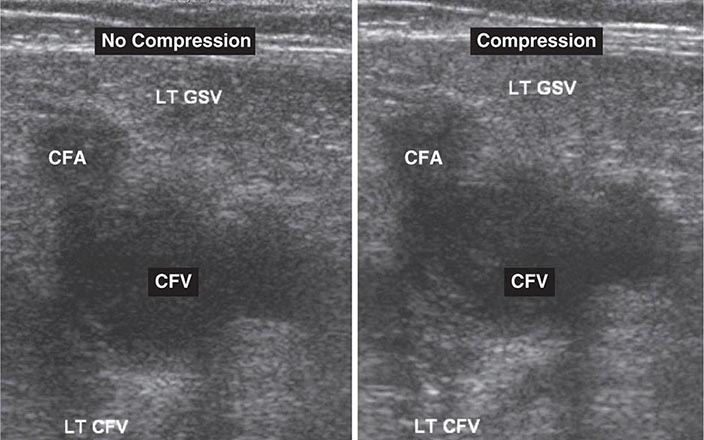
FIGURE 300-4 Venous ultrasound, with and without compression of the leg veins. CFA, common femoral artery; CFV, common femoral vein; GSV, great saphenous vein; LT, left.
Venous flow dynamics can be examined with Doppler imaging. Normally, manual calf compression causes augmentation of the Doppler flow pattern. Loss of normal respiratory variation is caused by an obstructing DVT or by any obstructive process within the pelvis. For patients with a technically poor or nondiagnostic venous ultrasound, one should consider alternative imaging modalities for DVT, such as computed tomography (CT) and magnetic resonance imaging.
CHEST ROENTGENOGRAPHY A normal or nearly normal chest x-ray often occurs in PE. Well-established abnormalities include focal oligemia (Westermark’s sign), a peripheral wedged-shaped density above the diaphragm (Hampton’s hump), and an enlarged right descending pulmonary artery (Palla’s sign).
CHEST CT CT of the chest with intravenous contrast is the principal imaging test for the diagnosis of PE (Fig. 300-5). Multidetector-row spiral CT acquires all chest images with ≤1 mm of resolution during a short breath hold. Sixth-order branches can be visualized with resolution superior to that of conventional invasive contrast pulmonary angiography. The CT scan also provides an excellent four-chamber view of the heart. RV enlargement on chest CT indicates an increased likelihood of death within the next 30 days compared with PE patients who have normal RV size. When imaging is continued below the chest to the knee, pelvic and proximal leg DVT also can be diagnosed by CT scanning. In patients without PE, the lung parenchymal images may establish alternative diagnoses not apparent on chest x-ray that explain the presenting symptoms and signs such as pneumonia, emphysema, pulmonary fibrosis, pulmonary mass, and aortic pathology. Sometimes asymptomatic early-stage lung cancer is diagnosed incidentally.
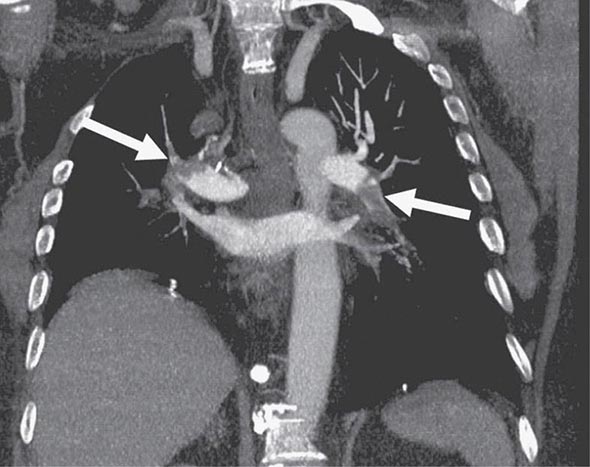
FIGURE 300-5 Large bilateral proximal PE on a coronal chest CT image in a 54-year-old man with lung cancer and brain metastases. He had developed sudden onset of chest heaviness and shortness of breath while at home. There are filling defects in the main and segmental pulmonary arteries bilaterally (white arrows). Only the left upper lobe segmental artery is free of thrombus.
LUNG SCANNING Lung scanning has become a second-line diagnostic test for PE, used mostly for patients who cannot tolerate intravenous contrast. Small particulate aggregates of albumin labeled with a gamma-emitting radionuclide are injected intravenously and are trapped in the pulmonary capillary bed. The perfusion scan defect indicates absent or decreased blood flow, possibly due to PE. Ventilation scans, obtained with a radiolabeled inhaled gas such as xenon or krypton, improve the specificity of the perfusion scan. Abnormal ventilation scans indicate abnormal nonventilated lung, thereby providing possible explanations for perfusion defects other than acute PE, such as asthma and chronic obstructive pulmonary disease. A high-probability scan for PE is defined as two or more segmental perfusion defects in the presence of normal ventilation.
The diagnosis of PE is very unlikely in patients with normal and nearly normal scans and is about 90% certain in patients with high-probability scans. Unfortunately, most patients have nondiagnostic scans, and fewer than one-half of patients with angiographically confirmed PE have a high probability scan. As many as 40% of patients with high clinical suspicion for PE but “low-probability” scans do, in fact, have PE at angiography.
MAGNETIC RESONANCE (MR) (CONTRAST-ENHANCED) IMAGING When ultrasound is equivocal, MR venography with gadolinium contrast is an excellent imaging modality to diagnose DVT. MR pulmonary angiography may detect large proximal PE but is not reliable for smaller segmental and subsegmental PE.
ECHOCARDIOGRAPHY Echocardiography is not a reliable diagnostic imaging tool for acute PE because most patients with PE have normal echocardiograms. However, echocardiography is a very useful diagnostic tool for detecting conditions that may mimic PE, such as acute myocardial infarction, pericardial tamponade, and aortic dissection. Transthoracic echocardiography rarely images thrombus directly. The best-known indirect sign of PE on transthoracic echocardiography is McConnell’s sign: hypokinesis of the RV free wall with normal or hyperkinetic motion of the RV apex. One should consider transesophageal echocardiography when CT scanning facilities are not available or when a patient has renal failure or severe contrast allergy that precludes administration of contrast despite premedication with high-dose steroids. This imaging modality can identify saddle, right main, or left main PE.
Invasive Diagnostic Modalities • PULMONARY ANGIOGRAPHY Chest CT with contrast (see above) has virtually replaced invasive pulmonary angiography as a diagnostic test. Invasive catheter-based diagnostic testing is reserved for patients with technically unsatisfactory chest CTs and for those in whom an interventional procedure such as catheter-directed thrombolysis is planned. A definitive diagnosis of PE depends on visualization of an intraluminal filling defect in more than one projection. Secondary signs of PE include abrupt occlusion (“cut-off”) of vessels, segmental oligemia or avascularity, a prolonged arterial phase with slow filling, and tortuous, tapering peripheral vessels.
CONTRAST PHLEBOGRAPHY Venous ultrasonography has virtually replaced contrast phlebography as the diagnostic test for suspected DVT.
Integrated Diagnostic Approach An integrated diagnostic approach (Fig. 300-3) streamlines the workup of suspected DVT and PE (Fig. 300-6).
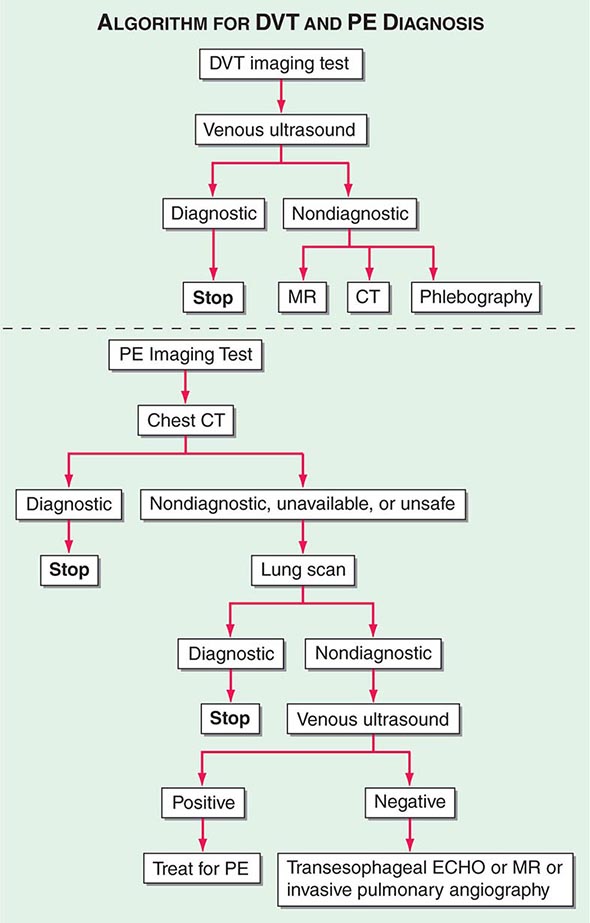
FIGURE 300-6 Imaging tests to diagnose DVT and PE. ECHO, echocardiography.
TREATMENT DEEP VENOUS THROMBOSIS
PRIMARY THERAPY
Primary therapy consists of clot dissolution with pharmacomechanical therapy that usually includes low-dose catheter-directed thrombolysis. This approach is reserved for patients with extensive femoral, iliofemoral, or upper extremity DVT. The open vein hypothesis postulates that patients who receive primary therapy will sustain less long-term damage to venous valves, with consequent lower rates of postthrombotic syndrome. A National Heart, Lung, and Blood Institute–sponsored randomized controlled trial called ATTRACT (NCT00790335) is testing this hypothesis.
SECONDARY PREVENTION
Anticoagulation or placement of an inferior vena caval filter constitutes secondary prevention of VTE. To lessen the severity of postthrombotic syndrome of the legs, below-knee graduated compression stockings may be prescribed, 30–40 mmHg, for 2 years after the DVT episode. They should be replaced every 3 months because they lose their elasticity.
TREATMENT PULMONARY EMBOLISM
RISK STRATIFICATION
Hemodynamic instability, RV dysfunction on echocardiography, RV enlargement on chest CT, or elevation of the troponin level due to RV microinfarction portend a high risk of an adverse clinical outcome. When RV function remains normal in a hemodynamically stable patient, a good clinical outcome is highly likely with anticoagulation alone (Fig. 300-7).
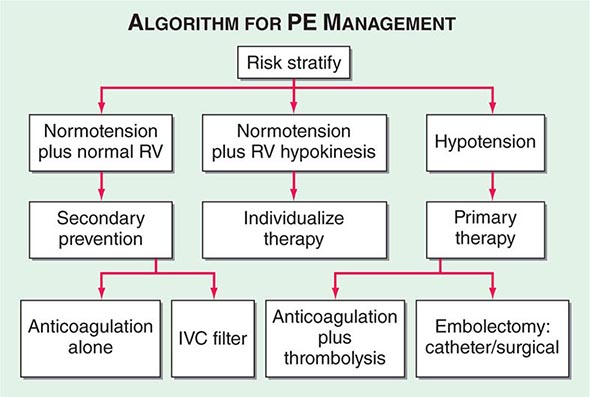
FIGURE 300-7 Acute management of pulmonary thromboembolism. RV, right ventricular; IVC, inferior vena cava.
ANTICOAGULATION
Effective anticoagulation is the foundation for successful treatment of DVT and PE. There are three options: (1) the conventional strategy of parenteral therapy “bridged” to warfarin, (2) parenteral therapy “bridged” to a novel oral anticoagulant such as dabigatran (a direct thrombin inhibitor) or edoxaban (an anti-Xa agent), or (3) oral anticoagulation with rivaroxaban or apixaban (both are anti-Xa agents) with a loading dose followed by a maintenance dose as monotherapy without parenteral anticoagulation.
The three heparin-based parenteral anticoagulants are (1) unfractionated heparin (UFH), (2) low-molecular-weight heparin (LMWH), and (3) fondaparinux. For patients with suspected or proven heparin-induced thrombocytopenia, there are two parenteral direct thrombin inhibitors: argatroban and bivalirudin (Table 300-3).
ANTICOAGULATION OF VTE |
Unfractionated Heparin UFH anticoagulates by binding to and accelerating the activity of antithrombin, thus preventing additional thrombus formation. UFH is dosed to achieve a target activated partial thromboplastin time (aPTT) of 60–80 s. The most popular nomogram uses an initial bolus of 80 U/kg, followed by an initial infusion rate of 18 U/kg per h.
The major advantage of UFH is its short half-life, which is especially useful in patients in whom hour-to-hour control of the intensity of anticoagulation is desired.
Low-Molecular-Weight Heparins These fragments of UFH exhibit less binding to plasma proteins and endothelial cells and consequently have greater bioavailability, a more predictable dose response, and a longer half-life than does UFH. No monitoring or dose adjustment is needed unless the patient is markedly obese or has chronic kidney disease.
Fondaparinux Fondaparinux, an anti-Xa pentasaccharide, is administered as a weight-based once-daily subcutaneous injection in a prefilled syringe. No laboratory monitoring is required. Fondaparinux is synthesized in a laboratory and, unlike LMWH or UFH, is not derived from animal products. It does not cause heparin-induced thrombocytopenia. The dose must be adjusted downward for patients with renal dysfunction.
Warfarin This vitamin K antagonist prevents carboxylation activation of coagulation factors II, VII, IX, and X. The full effect of warfarin requires at least 5 days, even if the prothrombin time, used for monitoring, becomes elevated more rapidly. If warfarin is initiated as monotherapy during an acute thrombotic illness, a paradoxical exacerbation of hypercoagulability increases the likelihood of thrombosis. Overlapping UFH, LMWH, fondaparinux, or parenteral direct thrombin inhibitors with warfarin for at least 5 days will nullify the early procoagulant effect of warfarin.
WARFARIN DOSING In an average-size adult, warfarin is often initiated in a dose of 5 mg. The prothrombin time is standardized by calculating the international normalized ratio (INR), which assesses the anticoagulant effect of warfarin (Chap. 78). The target INR is usually 2.5, with a range of 2.0–3.0.
The warfarin dose is usually titrated empirically to achieve the target INR. Proper dosing is difficult because hundreds of drug-drug and drug-food interactions affect warfarin metabolism. Increasing age and systemic illness reduce the required warfarin dose. Pharmacogenomics may provide more precise initial dosing of warfarin. CYP2C9 variant alleles impair the hydroxylation of S-warfarin, thereby lowering the dose requirement. Variants in the gene encoding the vitamin K epoxide reductase complex 1 (VKORC1) can predict whether patients require low, moderate, or high warfarin doses.
Centralized anticoagulation clinics have improved the efficacy and safety of warfarin dosing. Patients can self-monitor their INR with a home point-of-care fingerstick machine and can occasionally be taught to self-dose their warfarin.
Novel Oral Anticoagulants Novel oral anticoagulants are administered in a fixed dose, establish effective anticoagulation within hours of ingestion, require no laboratory coagulation monitoring, and have few of the drug-drug or drug-food interactions that make warfarin so difficult to dose. Rivaroxaban, a factor Xa inhibitor, is approved for treatment of acute DVT and acute PE as monotherapy, without a parenteral “bridging” anticoagulant. Apixaban is likely to receive similar approval for oral monotherapy. Dabigatran, a direct thrombin inhibitor, and edoxaban, a factor Xa inhibitor, are likely to be approved for treatment of VTE after an initial course of parenteral anticoagulation.
Complications of Anticoagulants The most serious adverse effect of anticoagulation is hemorrhage. For life-threatening or intracranial hemorrhage due to heparin or LMWH, protamine sulfate can be administered. Heparin-induced thrombocytopenia is less common with LMWH than with UFH. There is no specific reversal agent for bleeding caused by fondaparinux, direct thrombin inhibitors, or factor Xa inhibitors.
Major bleeding from warfarin is best managed with prothrombin complex concentrate. With serious but non–life-threatening bleeding, fresh-frozen plasma or intravenous vitamin K can be used. Recombinant human coagulation factor VIIa (rFVIIa) is an off-label option to manage catastrophic bleeding from warfarin, but prothrombin complex concentrate is a better choice. Oral vitamin K is effective for managing minor bleeding or an excessively high INR in the absence of bleeding.
Duration of Anticoagulation For DVT isolated to an upper extremity or calf that has been provoked by surgery, trauma, estrogen, or an indwelling central venous catheter or pacemaker, 3 months of anticoagulation usually suffice. For an initial episode of provoked proximal leg DVT or PE, 3 to 6 months of anticoagulation are considered sufficient. For patients with cancer and VTE, prescribe LMWH as monotherapy without warfarin and continue anticoagulation indefinitely unless the patient is rendered cancer-free.
Among patients with idiopathic, unprovoked VTE, the recurrence rate is high after cessation of anticoagulation. VTE that occurs during long-haul air travel is considered unprovoked. Unprovoked VTE may be caused by an exacerbation of an underlying inflammatory state and can be conceptualized as a chronic illness, with latent periods between flares of recurrent episodes. American College of Chest Physicians (ACCP) guidelines recommend considering anticoagulation for an indefinite duration with a target INR between 2 and 3 for patients with idiopathic VTE. An alternative approach after the first 6 months of anticoagulation is to reduce the intensity of anticoagulation and to lower the target INR range to between 1.5 and 2.
Counterintuitively, the presence of genetic mutations such as heterozygous factor V Leiden and prothrombin gene mutation does not appear to increase the risk of recurrent VTE. However, patients with antiphospholipid antibody syndrome may warrant indefinite-duration anticoagulation, even if the initial VTE was provoked by trauma or surgery.
INFERIOR VENA CAVAL (IVC) FILTERS
The two principal indications for insertion of an IVC filter are (1) active bleeding that precludes anticoagulation and (2) recurrent venous thrombosis despite intensive anticoagulation. Prevention of recurrent PE in patients with right heart failure who are not candidates for fibrinolysis and prophylaxis of extremely high-risk patients are “softer” indications for filter placement. The filter itself may fail by permitting the passage of small-to medium-size clots. Large thrombi may embolize to the pulmonary arteries via collateral veins that develop. A more common complication is caval thrombosis with marked bilateral leg swelling.
Paradoxically, by providing a nidus for clot formation, filters increase the DVT rate, even though they usually prevent PE (over the short term). Retrievable filters can now be placed for patients with an anticipated temporary bleeding disorder or for patients at temporary high risk of PE, such as individuals undergoing bariatric surgery who have a prior history of perioperative PE. The filters can be retrieved up to several months after insertion unless thrombus forms and is trapped within the filter. The retrievable filter becomes permanent if it remains in place or if, for technical reasons such as rapid endothelialization, it cannot be removed.
MANAGEMENT OF MASSIVE PE
For patients with massive PE and hypotension, replete volume with 500 mL of normal saline. Additional fluid should be infused with extreme caution because excessive fluid administration exacerbates RV wall stress, causes more profound RV ischemia, and worsens LV compliance and filling by causing further interventricular septal shift toward the LV. Dopamine and dobutamine are first-line inotropic agents for treatment of PE-related shock. Maintain a low threshold for initiating these pressors. Often, a “trial-and-error” approach works best; other agents that may be effective include norepinephrine, vasopressin, or phenylephrine.
FIBRINOLYSIS
Successful fibrinolytic therapy rapidly reverses right heart failure and may result in a lower rate of death and recurrent PE by (1) dissolving much of the anatomically obstructing pulmonary arterial thrombus, (2) preventing the continued release of serotonin and other neurohumoral factors that exacerbate pulmonary hypertension, and (3) lysing much of the source of the thrombus in the pelvic or deep leg veins, thereby decreasing the likelihood of recurrent PE.
The preferred fibrinolytic regimen is 100 mg of recombinant tissue plasminogen activator (tPA) administered as a continuous peripheral intravenous infusion over 2 h. The sooner thrombolysis is administered, the more effective it is. However, this approach can be used for at least 14 days after the PE has occurred.
Contraindications to fibrinolysis include intracranial disease, recent surgery, and trauma. The overall major bleeding rate is about 10%, including a 1–3% risk of intracranial hemorrhage. Careful screening of patients for contraindications to fibrinolytic therapy (Chap. 295) is the best way to minimize bleeding risk.
The only Food and Drug Administration–approved indication for PE fibrinolysis is massive PE. For patients with submassive PE, who have preserved systolic blood pressure but moderate or severe RV dysfunction, use of fibrinolysis remains controversial. Results of a 1006-patient European multicentered randomized trial of submassive PE, using the thrombolytic agent tenecteplase, were published in 2014. Death or hemodynamic collapse within 7 days of randomization was reduced by 56% in the tenecteplase group. However, hemorrhagic stroke occurred in 2% of tenecteplase patients versus 0.2% in patients who only received heparin.
PHARMACOMECHANICAL CATHETER-DIRECTED THERAPY
Many patients have relative contraindications to full-dose thrombolysis. Pharmacomechanical catheter-directed therapy usually combines physical fragmentation or pulverization of thrombus with catheter-directed low-dose thrombolysis. Mechanical techniques include catheter maceration and intentional embolization of clot more distally, suction thrombectomy, rheolytic hydrolysis, and low-energy ultrasound-facilitated thrombolysis. The dose of alteplase can be markedly reduced, usually to a range of 20 to 25 mg instead of the peripheral intravenous systemic dose of 100 mg.
PULMONARY EMBOLECTOMY
The risk of major hemorrhage with systemically administered fibrinolysis has prompted a renaissance of interest in surgical embolectomy, an operation that had almost become extinct. More rapid referral before the onset of irreversible multisystem organ failure and improved surgical technique have resulted in a high survival rate.
PULMONARY THROMBOENDARTERECTOMY
Chronic thromboembolic pulmonary hypertension develops in 2–4% of acute PE patients. Therefore, PE patients who have initial pulmonary hypertension (usually diagnosed with Doppler echocardiography) should be followed up at about 6 weeks with a repeat echocardiogram to determine whether pulmonary arterial pressure has normalized. Patients impaired by dyspnea due to chronic thromboembolic pulmonary hypertension should be considered for pulmonary thromboendarterectomy, which, if successful, can markedly reduce, and sometimes even cure, pulmonary hypertension (Chap. 304). The operation requires median sternotomy, cardiopulmonary bypass, deep hypothermia, and periods of hypothermic circulatory arrest. The mortality rate at experienced centers is approximately 5%. Inoperable patients should be managed with pulmonary vasodilator therapy.
EMOTIONAL SUPPORT
Patients with VTE may feel overwhelmed when they learn that they are suffering from PE or DVT. Some have never previously encountered serious cardiovascular illness. They wonder whether they will be able to adapt to the new limitations imposed by anticoagulation. They worry about the health of their families and the genetic implications of their illness. Those who are advised to discontinue anticoagulation may feel especially vulnerable about the potential for suffering recurrent VTE. At Brigham and Woman’s Hospital, a physician-nurse–facilitated PE support group was initiated to address these concerns and has met monthly for more than 20 years.
PREVENTION OF VTE
Prevention of DVT and PE (Table 300-4) is of paramount importance because VTE is difficult to detect and poses a profound medical and economic burden. Low-dose UFH or LMWH is the most common form of in-hospital prophylaxis. Computerized reminder systems can increase the use of preventive measures and, at Brigham and Women’s Hospital, have reduced the symptomatic VTE rate by more than 40%. Audits of hospitals to ensure that prophylaxis protocols are being used will also increase utilization of preventive measures. Duration of prophylaxis is an important consideration. Extended-duration prophylaxis has not been shown to be both effective and safe in medically ill patients after hospital discharge in separate large trials that have tested enoxaparin, apixaban, and rivaroxaban. There is an ongoing trial of a novel oral anticoagulant, betrixaban, for extended-duration VTE prophylaxis in medically ill patients.
PREVENTION OF VENOUS THROMBOEMBOLISM AMONG HOSPITALIZED PATIENTS |
Patients who have undergone total hip or knee replacement or cancer surgery will benefit from extended pharmacologic VTE prophylaxis after hospital discharge. For hip replacement or extensive cancer surgery, the duration of prophylaxis is usually at least 1 month.
301 | Diseases of the Aorta |
The aorta is the conduit through which blood ejected from the left ventricle is delivered to the systemic arterial bed. In adults, its diameter is approximately 3 cm at the origin and in the ascending portion, 2.5 cm in the descending portion in the thorax, and 1.8–2 cm in the abdomen. The aortic wall consists of a thin intima composed of endothelium, subendothelial connective tissue, and an internal elastic lamina; a thick tunica media composed of smooth muscle cells and extracellular matrix; and an adventitia composed primarily of connective tissue enclosing the vasa vasorum and nervi vascularis. In addition to the conduit function of the aorta, its viscoelastic and compliant properties serve a buffering function. The aorta is distended during systole to allow a portion of the stroke volume and elastic energy to be stored, and it recoils during diastole so that blood continues to flow to the periphery. Owing to its continuous exposure to high pulsatile pressure and shear stress, the aorta is particularly prone to injury and disease resulting from mechanical trauma. The aorta is also more prone to rupture than is any other vessel, especially with the development of aneurysmal dilation, since its wall tension, as governed by Laplace’s law (i.e., proportional to the product of pressure and radius), will be increased.
CONGENITAL ANOMALIES OF THE AORTA
Congenital anomalies of the aorta usually involve the aortic arch and its branches. Symptoms such as dysphagia, stridor, and cough may occur if an anomaly causes a ring around or otherwise compresses the esophagus or trachea. Anomalies associated with symptoms include double aortic arch, origin of the right subclavian artery distal to the left subclavian artery, and right-sided aortic arch with an aberrant left subclavian artery. A Kommerell’s diverticulum is an anatomic remnant of a right aortic arch. Most congenital anomalies of the aorta do not cause symptoms and are detected during catheter-based procedures. The diagnosis of suspected congenital anomalies of the aorta typically is confirmed by computed tomographic (CT) or magnetic resonance (MR) angiography. Surgery is used to treat symptomatic anomalies.
AORTIC ANEURYSM
An aneurysm is defined as a pathologic dilation of a segment of a blood vessel. A true aneurysm involves all three layers of the vessel wall and is distinguished from a pseudoaneurysm, in which the intimal and medial layers are disrupted and the dilated segment of the aorta is lined by adventitia only and, at times, by perivascular clot. Aneurysms also may be classified according to their gross appearance. A fusiform aneurysm affects the entire circumference of a segment of the vessel, resulting in a diffusely dilated artery. In contrast, a saccular aneurysm involves only a portion of the circumference, resulting in an outpouching of the vessel wall. Aortic aneurysms also are classified according to location, i.e., abdominal versus thoracic. Aneurysms of the descending thoracic aorta are usually contiguous with infradiaphragmatic aneurysms and are referred to as thoracoabdominal aortic aneurysms.
ETIOLOGY
Aortic aneurysms result from conditions that cause degradation or abnormal production of the structural components of the aortic wall: elastin and collagen. The causes of aortic aneurysms may be broadly categorized as degenerative disorders, genetic or developmental diseases, vasculitis, infections, and trauma (Table 301-1). Inflammation, oxidative stress, proteolysis, and biomechanical wall stress contribute to the degenerative processes that characterize most aneurysms of the abdominal and descending thoracic aorta. These are mediated by B cell and T cell lymphocytes, macrophages, inflammatory cytokines, and matrix metalloproteinases that degrade elastin and collagen and alter the tensile strength and ability of the aorta to accommodate pulsatile stretch. The associated histopathology demonstrates destruction of elastin and collagen, decreased vascular smooth muscle, in-growth of new blood vessels, and inflammation. Factors associated with degenerative aortic aneurysms include aging, cigarette smoking, hypercholesterolemia, hypertension, and male sex.
DISEASES OF THE AORTA: ETIOLOGY AND ASSOCIATED FACTORS |
The most common pathologic condition associated with degenerative aortic aneurysms is atherosclerosis. Many patients with aortic aneurysms have coexisting risk factors for atherosclerosis (Chap. 291e), as well as atherosclerosis in other blood vessels.
Medial degeneration, previously designated cystic medial necrosis, is the histopathologic term used to describe the degeneration of collagen and elastic fibers in the tunica media of the aorta as well as the loss of medial cells that are replaced by multiple clefts of mucoid material, such as proteoglycans. Medial degeneration characteristically affects the proximal aorta, results in circumferential weakness and dilation, and leads to the development of fusiform aneurysms involving the ascending aorta and the sinuses of Valsalva. This condition is particularly prevalent in patients with Marfan’s syndrome, Loeys-Dietz syndrome, Ehlers-Danlos syndrome type IV (Chap. 427), hypertension, congenital bicuspid aortic valves, and familial thoracic aortic aneurysm syndromes; sometimes it appears as an isolated condition in patients without any other apparent disease.
Familial clusterings of aortic aneurysms occur in 20% of patients, suggesting a hereditary basis for the disease. Mutations of the gene that encodes fibrillin-1 are present in patients with Marfan’s syndrome. Fibrillin-1 is an important component of extracellular microfibrils, which support the architecture of elastic fibers and other connective tissue. Deficiency of fibrillin-1 in the extracellular matrix leads to excessive signaling by transforming growth factor β (TGF-β). Loeys-Dietz syndrome is caused by mutations in the genes that encode TGF-β receptors 1 (TGFBR1) and 2 (TGFBR2). Increased signaling by TGF-β and mutations of TGFBR1 and TGFBR2 may cause thoracic aortic aneurysms. Mutations of type III procollagen have been implicated in Ehlers-Danlos type IV syndrome. Mutations of SMAD3, which encodes a downstream signaling protein involved with TGF binding to its receptors, have been described in a syndrome of thoracic aortic aneurysm; craniofacial, skeletal, and cutaneous anomalies; and osteoarthritis. Mutations of the genes encoding the smooth muscle–specific alpha-actin (ACTA2), smooth muscle cell–specific myosin heavy chain 11 (MHC11), and myosin light chain kinase (MYLK) and mutations of TGFBR2 and SMAD3 have been reported in some patients with nonsyndromic familial thoracic aortic aneurysms.
The infectious causes of aortic aneurysms include syphilis, tuberculosis, and other bacterial infections. Syphilis (Chap. 206) is a relatively uncommon cause of aortic aneurysm. Syphilitic periaortitis and mesoaortitis damage elastic fibers, resulting in thickening and weakening of the aortic wall. Approximately 90% of syphilitic aneurysms are located in the ascending aorta or aortic arch. Tuberculous aneurysms (Chap. 202) typically affect the thoracic aorta and result from direct extension of infection from hilar lymph nodes or contiguous abscesses as well as from bacterial seeding. Loss of aortic wall elasticity results from granulomatous destruction of the medial layer. A mycotic aneurysm is a rare condition that develops as a result of staphylococcal, streptococcal, Salmonella, or other bacterial or fungal infections of the aorta, usually at an atherosclerotic plaque. These aneurysms are usually saccular. Blood cultures are often positive and reveal the nature of the infective agent.
Vasculitides associated with aortic aneurysm include Takayasu’s arteritis and giant cell arteritis, which may cause aneurysms of the aortic arch and descending thoracic aorta. Spondyloarthropathies such as ankylosing spondylitis, rheumatoid arthritis, psoriatic arthritis, relapsing polychondritis, and reactive arthritis (formerly known as Reiter’s syndrome) are associated with dilation of the ascending aorta. Aortic aneurysms occur in patients with Behçet’s syndrome (Chap. 387), Cogan’s syndrome, and IgG4-related systemic disease. Aortic aneurysms also result from idiopathic aortitis. Traumatic aneurysms may occur after penetrating or nonpenetrating chest trauma and most commonly affect the descending thoracic aorta just beyond the site of insertion of the ligamentum arteriosum. Chronic aortic dissections are associated with weakening of the aortic wall that may lead to the development of aneurysmal dilatation.
THORACIC AORTIC ANEURYSMS
The clinical manifestations and natural history of thoracic aortic aneurysms depend on their location. Medial degeneration is the most common pathology associated with ascending aortic aneurysms, whereas atherosclerosis is the condition most frequently associated with aneurysms of the descending thoracic aorta. The average growth rate of thoracic aneurysms is 0.1–0.2 cm per year. Thoracic aortic aneurysms associated with Marfan’s syndrome or aortic dissection may expand at a greater rate. The risk of rupture is related to the size of the aneurysm and the presence of symptoms, ranging approximately from 2–3% per year for thoracic aortic aneurysms <4.0 cm in diameter to 7% per year for those >6 cm in diameter. Most thoracic aortic aneurysms are asymptomatic; however, compression or erosion of adjacent tissue by aneurysms may cause symptoms such as chest pain, shortness of breath, cough, hoarseness, and dysphagia. Aneurysmal dilation of the ascending aorta may cause congestive heart failure as a consequence of aortic regurgitation, and compression of the superior vena cava may produce congestion of the head, neck, and upper extremities.
A chest x-ray may be the first test that suggests the diagnosis of a thoracic aortic aneurysm (Fig. 301-1). Findings include widening of the mediastinal shadow and displacement or compression of the trachea or left main stem bronchus. Echocardiography, particularly transesophageal echocardiography, can be used to assess the proximal ascending aorta and descending thoracic aorta. Contrast-enhanced CT, magnetic resonance imaging (MRI), and conventional invasive aortography are sensitive and specific tests for assessment of aneurysms of the thoracic aorta and involvement of branch vessels (Fig. 301-2). In asymptomatic patients whose aneurysms are too small to justify surgery, noninvasive testing with either contrast-enhanced CT or MRI should be performed at least every 6–12 months to monitor expansion.
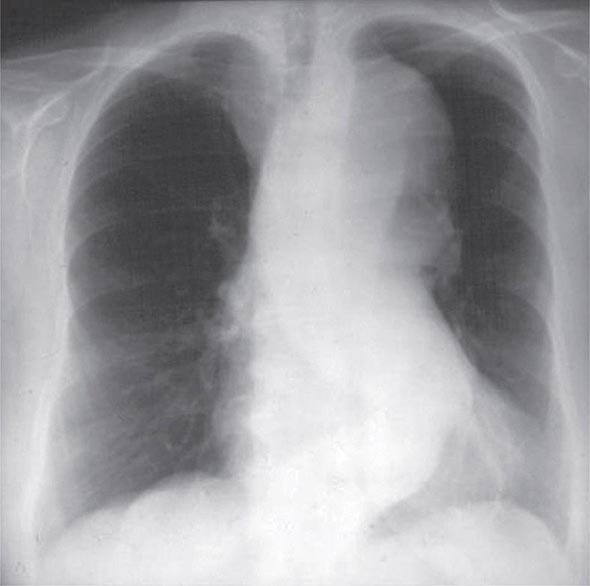
FIGURE 301-1 A chest x-ray of a patient with a thoracic aortic aneurysm.
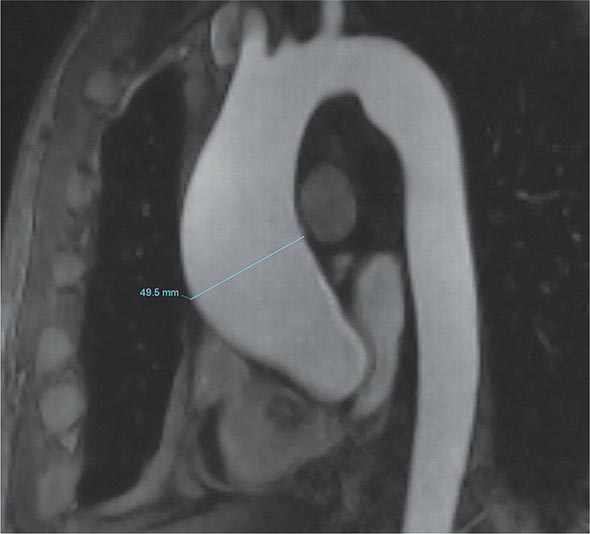
FIGURE 301-2 A magnetic resonance angiogram demonstrating a fusiform aneurysm of the ascending thoracic aorta. (Courtesy of Dr. Michael Steigner, Brigham and Women’s Hospital, Boston, MA, with permission.)
TREATMENT THORACIC AORTIC ANEURYSMS
β-Adrenergic blockers currently are recommended for patients with thoracic aortic aneurysms, particularly those with Marfan’s syndrome, who have evidence of aortic root dilatation to reduce the rate of further expansion. Additional medical therapy should be given as necessary to control hypertension. Recent studies indicate that angiotensin receptor antagonists and angiotensin-converting enzyme inhibitors reduce the rate of aortic dilation in patients with Marfan’s syndrome by blocking TGF-β signaling; clinical outcome trials of this treatment approach are in progress. Operative repair with placement of a prosthetic graft is indicated in patients with symptomatic ascending thoracic aortic aneurysms and for most asymptomatic aneurysms when the ascending aortic diameter is >5.5 cm. In patients with Marfan’s syndrome or bicuspid aortic valve, ascending thoracic aortic aneurysms of 4–5 cm should be considered for surgery. Operative repair is indicated for patients with descending thoracic aortic aneurysms when the diameter is >6 cm, and endovascular repair should be considered if feasible when the diameter is >5.5 cm. Repair is also recommended when the diameter of an aneurysm has increased >1 cm per year.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


