- Daptomycin is approved for the treatment of MRSA-linked complicated skin and skin-structure infections (cSSSIs) and bacteraemia, including right-sided endocarditis.
- The mode of action is not well understood, but involves disruption of the cell wall of Gram positive bacteria after insertion of daptomycin, facilitated by its lipophilic ‘tail’.
- Daptomycin was only recently introduced and resistance is not yet common; to avoid the development of resistance, it should be used rarely and only for the indications above.
Cubicin (daptomycin for injection) was approved by the FDA in 2003 for the treatment of complicated SSSIs caused by MRSA, and in 2006 the FDA approved a new indication: the treatment of S. aureus bacteraemia due to MRSA, including right-sided endocarditis.
5.5.1 Discovery
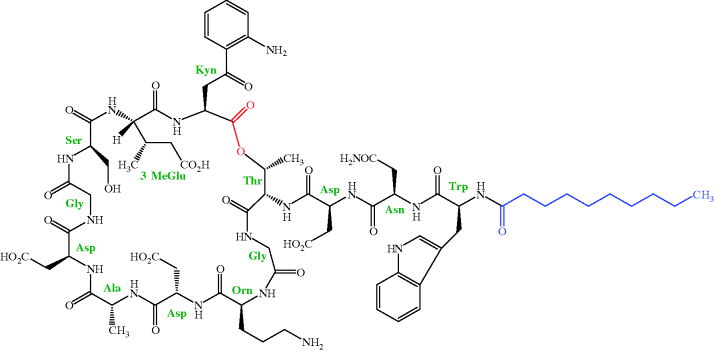
As mentioned in Section 1, daptomycin is one of only two truly novel antibacterial agents launched in the last 30 years. Daptomycin is a lipopeptide and was originally isolated by researchers at Eli Lilly and Co. from a strain of Streptomyces roseosporus in a soil sample from Mount Ararat21 in Turkey. As you can see from the structure in Figure 5.5.1, daptomycin contains 13 amino acids (not all of which are proteinogenic), 10 of which form a depsipeptide22 ring, with the other three attached to this ring through a threonine residue. This depsipeptide forms the core of the family of lipopeptides (called A21978C) isolated from this organism. The 13-amino-acid core is common to all members of the A21978C complex and it is only the lipid portion which differs, with the fatty acid side chain being n-decanoyl (C10) in daptomycin (which was originally called LY 146032), a minor component of the mixture. Once the antibacterial activity of these lipopeptides had been determined, daptomycin was the agent chosen for clinical development as this fatty acid side chain was associated with the highest in vivo efficacy and lowest toxicity in animals (Eisenstein et al., 2010). As you might imagine, all the possible fatty acid derivatives of the core peptide (as well as some containing other functional groups) were prepared and evaluated for their antibacterial activity, with only a few being selected for further evaluation. Amazingly, even a one-carbon difference in the fatty acid chain was found to lead to a dramatic increase in toxicity, as when mice were dosed with 1 g/kg (IV) of the n-undecanoyl (C11) derivative, 100% were killed, compared to 20% for the n-decanoyl (C10) analogue (daptomycin).
For more on the discovery of daptomycin, see Tally and DeBruin (2000), Baltz et al. (2005), and Eisenstein et al. (2010).
5.5.2 Synthesis
As is the case for the other very complex antibiotics discussed throughout this book, the total synthesis of daptomycin is not a viable option for the manufacture of sufficient quantities of this drug for clinical use. Daptomycin can be produced by one of two methods, both of which have some similarity to the synthesis of the penicillins (which we covered in Section 5.1). Eli Lilly, the company that first isolated daptomycin, developed a method which converted the A21978C complex to the core peptide (using a deacylase enzyme isolated from Actinoplanes utahensis NRRL 12052), which could then be re-acylated with different lipid side chains (Scheme 5.5.1) (DeBono et al., 1988). As shown in Scheme 5.5.1, this process is not as straightforward as you might expect and involves a couple more steps than you might have anticipated. The de-acylated intermediate is re-acylated with an active ester (in this case, the 2,3,5-trichlorophenyl ester of decanoic acid) to give daptomycin.
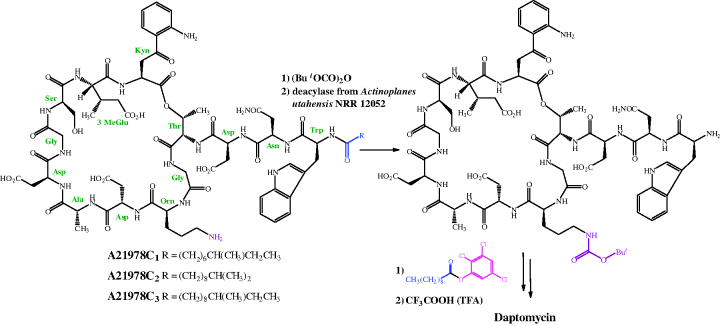
As it involves four steps, some of which give yields in the 50–60% range, the de-acylation/re-acylation method was not deemed to be cost-effective for full-scale manufacture of daptomycin, so an alternative method was developed, in which decanoic acid was fed into the fermentation cultures of S. roseosporus, thus producing daptomycin directly (Huber et al., 1988).
5.5.3 Bioavailability
Daptomycin is administered intravenously and has an apparent volume of distribution of 0.1 L/kg, which may be affected by a high degree of plasma protein binding (90–95%). It has an elimination half-life of approximately 9 hours and, as it is eliminated unchanged by renal excretion (approx. 54%), the dose interval should be increased to 48 hours in patients with impaired renal function. In a study conducted in 24 healthy male and female volunteers treated with daptomycin for 7–14 days, it showed linear pharmacokinetics and, at 4 mg/kg, had a mean Cmax of 54.6 μg/mL and an AUC of 494 mg/hour/mL in patients treated for 7 days (Dvorchik et al., 2003).
As daptomycin interacts with, and inserts into, the lipid cell wall of Gram positive bacteria, it does not have to cross the entire membrane or enter the cell, only requiring access through the outer components of the membrane. Gram positive bacteria have a more readily traversed outer membrane, while the Gram negative bacteria are resistant due to their more complex membrane structure.
For more on the bioavailability of daptomycin, see Jeu and Fung (2004).
5.5.4 Mode of Action and Selectivity
You are probably thinking that, as daptomycin was discovered relatively recently and approved for use only within the last 10 years, its mode of action will have been the subject of extensive research investigations and so will be fully understood. Surprisingly, especially for a drug which is in clinical use, the mode of action of daptomycin is by no means completely understood, and so we can only mention here what the evidence so far suggests.
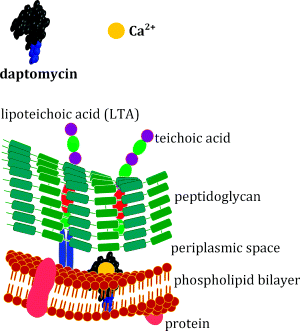
What is clear about the daptomycin mode of action is that calcium ions are essential for its rapid bactericidal activity against Gram positive bacteria (Steenbergen et al., 2005), which is diminished in the presence of alternative divalent ions. It was initially suggested that daptomycin interferes with lipoteichoic acid (LTA) synthesis, as it inhibits biosynthesis of LTA in Enterococcus hirae before that of other macromolecules (which gives it kinetic specificity) (Canepari et al., 1990; Boaretti et al., 1993). In this organism, daptomycin also exhibits dose-dependent effects, inhibiting only LTA synthesis at doses near the MIC, but multiple pathways at higher doses. As we saw in Section 1 (Figure 1.1.7), LTA is a cell-surface macromolecule which is anchored to the cytoplasmic membrane and extends into the peptidoglycan layer of the cell wall. The dose and kinetic specificity exhibited by daptomycin in E. hirae were not mirrored in Staphylococcus aureus, suggesting that a different mode of action is operating, and this is consistent with the fact that LTAs show marked structural diversity among Gram positive bacteria, meaning that it is unlikely that they would function as a single target for daptomycin.
The mode of action of daptomycin in S. aureus or Enterococcus faecalis is currently believed to involve the insertion of daptomycin into the lipid bilayer (Figure 5.5.2) (Laganas et al., 2003; Silverman et al., 2003). This insertion is facilitated by the lipid tail of the antibiotic, which initially promotes weak hydrophobic interactions with the lipids of the phospholipid bilayer. What happens before this insertion into the lipid bilayer, and how daptomycin inserting into the membrane leads to cell death, is not yet entirely clear. One study has shown that daptomycin experiences two structural transitions on contact with a cell membrane (Jung et al., 2004). First, a structural transition occurs with the simple interaction of daptomycin and calcium itself, followed by an interaction with calcium and the phosphatidyl glycerol. These interactions promote mild disturbances in the lipid membrane and cause content leakage. This allows interaction with the cytoplasmic membrane and alters permeability with enhanced daptomycin-membrane adhesion. It is believed that daptomycin oligomerisation23 (which is promoted by binding to Ca2+) creates a large pore in the membrane, which allows potassium efflux, membrane depolarisation, and eventually cell death (Laganas et al., 2003; Silverman et al., 2003). This mode of action is consistent with the fact that daptomycin has reduced in vivo activity against Streptococcus pneumoniae, despite having potent bactericidal activity against this organism in vitro. In the lungs, daptomycin is thought to interact with pulmonary surfactant (a component of epithelial lining fluid which consists primarily of the phospholipid dipalmitoylphosphatidylcholine) in preference to the phospholipid bilayer of these bacteria (Silverman et al., 2005).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


