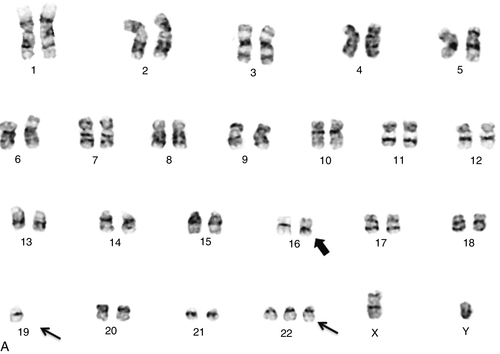
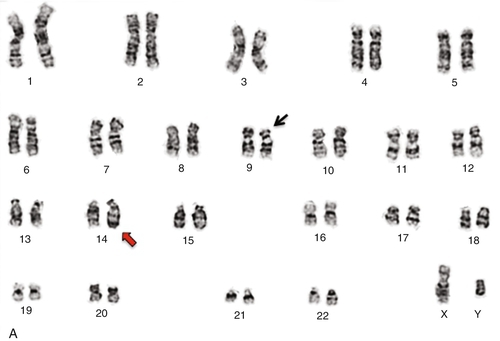
Chapter 23 Vundavalli V. Murty; Vaidehi Jobanputra; Brynn Levy B. 45,XY,der(13;21)(q10;q10),+21. C. 47,XY,+mar. D. 46,XY,del(7)(q11.2;q11.2). E. 46,XY,add(11)(q23). 2. A bone marrow specimen was submitted for cytogenetics analysis from a 3-month-old boy presenting with a white blood cell count of 289,700/μL, a hemoglobin of 6.4 g/dL, and a platelet count of 17,000/μL. The bone marrow aspirate smear was highly cellular with many blasts. Chromosome analysis showed simple karyotypic changes (Figure 23-1, A). The karyotype is described as 46,XY,t(2;19)(q23;q13.4),t(4;11)(q21;q23). Based on the breakpoint at 11q23, fluorescence in situ hybridization (FISH) analysis was performed using a myeloid/lymphoid leukemia (MLL) gene dual-color break-apart probe (Figure 23-1, B). The MLL gene was rearranged with the 4q21 locus, consistent with t(4;11)(q21; q23). Based on this karyotype and FISH analysis, which statement represents the most likely diagnosis and prognosis? A. Burkitt lymphoma (BL) with a poor prognosis. B. Myelodysplastic syndrome (MDS) with a good prognosis. C. Acute lymphoblastic leukemia (ALL) with a poor prognosis. D. ALL with a good prognosis. E. Acute promyelocytic leukemia (APL) with a good prognosis. 3. A bone marrow sample from a 13-year-old boy was submitted for cytogenetic workup to rule out non-Hodgkin lymphoma (NHL). The bone marrow aspirate showed a marked increase in lymphoid cells with sizes ranging from small to large, containing cleaved nuclei with multiple nucleoli. These abnormal cells had a coarsely reticular to fine chromatin and a small-to-moderate amount of blue-gray agranular cytoplasm. Flow cytometric analysis, after preferential gating for CD45 bright cells, showed a predominant lymphoid population that expresses CD45, CD16/56, CD7, CD13, HLA-DR, CD30, and CD25. No CD43, or surface or cytoplasmic CD3, or αβ or γδ T-cell receptors (TCRs) were identified in these atypical lymphocytes. Chromosome analysis (Figure 23-2, A) showed a simple karyotype consisting of a balanced translocation with breakpoints at chromosome regions 2p23 and 5q34, resulting in the t(2;5)(p23;q35) translocation (Figure 23-2, B) and an abnormal chromosome 8. FISH analysis using an anaplastic lymphoma kinase (ALK) break-apart probe was positive for rearrangement (Figure 23-2, C). Based on the morphologic, flow cytometric, and cytogenetic findings, what is the most likely diagnosis in this case? A. Natural killer (NK)-cell leukemia and lymphoma. B. Anaplastic large cell lymphoma (ALCL). C. Hepatosplenic T-cell lymphoma. D. T-cell prolymphocytic leukemia (T-PLL). E. Angioimmunoblastic T-cell lymphoma. 4. A bone marrow specimen from a 6-year-old boy with suspected acute leukemia was submitted for chromosome analysis. Histological examination of the bone marrow biopsy revealed complete replacement of normal cells with small to medium-sized immature blast cells (86%) with high mitotic activity. Flow cytometry analysis of the marrow in the CD45-negative gate showed immature precursor cells positive for CD19, HLA-DR, CD34, CD10, TdT, and CD22. Chromosome analysis showed loss of one copy of most of the autosomes, as shown in the Figure 23-3. This karyotype is described as 27 < 1 N >,XY,+14,+18,+21. Based on this karyotype, combined with the morphology and flow cytometry findings, which one of the following statements describes the most likely diagnosis and prognosis in this case? A. BL, predicting a poor prognosis. B. MDS, predicting a good prognosis. C. ALL, predicting a poor prognosis. D. ALL, predicting a good prognosis. E. APL, predicting a good prognosis. 5. Bone marrow biopsy from a 40-year-old man with acute myeloid leukemia (AML) shows a preponderance of hypergranular promyelocytes and bundles of Auer rods. Which one of the following is the most likely karyotype in his bone marrow metaphase cells? B. 46,XY,inv(16)(p13q22). C. 46,XY,t(4;11)(q21;q23). D. 46,XY,t(15;17)(q24;q21). E. 46,XY,inv(3)(q21q26). 6. A 13-year-old boy presented with a previous history of an autologous hematopoietic stem cell transplant as therapy for BL and currently has a mildly hypocellular bone marrow. Immunohistochemical staining was positive for CD19, negative for CD34, and positive for Epstein-Barr virus. The bone marrow specimen was submitted for cytogenetic workup. Chromosome analysis (Figure 23-4, A) showed a complex karyotype that included a translocation between chromosome regions 14q32 and 8q24, resulting in t(8;14), a duplication of 1q, and other structural abnormalities. FISH analysis using an IGH/MYC translocation probe confirmed the t(8;14) translocation (Figure 23-4, B). Based on this karyotype, what is the most likely diagnosis of this lymphoma? B. Hodgkin disease. C. Multiple myeloma. D. Mucosa-associated lymphoid tissue (MALT) lymphoma. E. Marginal zone lymphoma (MZL). 7. A 66-year-old woman with a history of end-stage renal disease (ESRD) treated with a renal transplant 3 years previously currently presented with acute renal failure and thrombocytopenia. A bone aspirate showed trilinear hematopoiesis with maturation and extensive marrow involvement by abnormal lymphoid cells. Immunohistochemical staining showed that this population was CD20+, CD10+, and CD43–. Flow cytometric analysis of the bone marrow showed the abnormal population to be CD10+, CD19+, CD20+, CD38+, CD5–, sIgM+, and λ-restricted. The bone marrow specimen was submitted for cytogenetic workup. Chromosome analysis (Figure 23-5, A) showed a translocation between chromosome regions 8q24 and 22q11.2, resulting in a t(8;22) translocation. FISH analysis using MYC and IGL break-apart probes were positive (Figure 23-5, C and D), confirming the t(8;22) translocation. Based on the morphologic, immunohistochemical, flow cytometric, and cytogenetic observations in this case, what is the most likely diagnosis? B. MZL. C. Multiple myeloma. D. MALT lymphoma. E. AML. 8. A blood sample from a patient diagnosed with chronic lymphocytic leukemia (CLL) was submitted for cytogenetic workup. Based on the karyotype and FISH images shown in Figure 23-6, and the corresponding FISH data, which one of the following interpretations is correct regarding the prognosis of CLL in this patient? A. The karyotype predicts a good prognosis. B. The two copies of the ataxia-telangiectasia mutated (ATM) gene by interphase FISH predict a good prognosis. C. The G-band karyotype and FISH together do not predict any particular prognosis. D. Heterozygous deletion of the TP53 gene predicts a poor prognosis. E. Heterozygous deletion of the TP53 gene predicts a good prognosis. 9. What is the best estimate of the incidence of chromosome abnormalities in spontaneous abortions according to recent cytogenetic studies on the products of conception from first-trimester miscarriages? B. 15% to 30%. C. 30% to 45%. D. 60% to 70%. E. 75% to 80%. 10. Which statement best describes how cytogenetic analysis routinely identifies chromosomes? A. Their specific bar code, the size of the centromere, and the amount of heterochromatin. B. The size of the chromosome, the banding pattern, and the length of the telomere. C. The length of the telomere, the location of the centromere, and their specific bar code. D. The position of the centromere, the size of the chromosome, and the banding pattern. E. The location of the heterochromatin, their specific bar code pattern, and the location of the centromere. 11. Which essential amino acid needs to be added to cell culture medium just prior to use? B. l-Lysine. C. l-Valine. D. l-Leucine. E. l-Glutamine. 12. Chromosome analysis, performed on a patient’s products of conception (POC), shows 10 cells with a 69,XXY karyotype and 10 cells with a 46,XX karyotype. Which is the most appropriate interpretation of this result? B. The 69,XXY cells are an artifact of tissue culture and the POC contains only normal female cells. C. The POC has a 69,XXY karyotype and the cells with the 46,XX karyotype are maternal in origin. D. This is a twin pregnancy: One twin has a 69,XXY karyotype and the other twin has a 46,XX karyotype. E. This represents true triploidy mosaicism; the fetus has two cell lines (69,XXY and 46,XX). 13. A bone marrow sample from a 53-year-old male patient with a history of anemia, leukocytosis, and thrombocytosis was submitted for cytogenetic workup. Morphologic analysis of a bone marrow core biopsy showed a myeloproliferative neoplasm with myelofibrosis. Flow cytometric analysis showed a phenotype of CD34+, HLA-DR+, CD117+, CD13+/–, and CD33+/– in 1% of the nucleated cells. Based on these observations, a diagnosis of a low-grade myeloid neoplasm was rendered. Karyotype analysis identified a deletion of the long arm of chromosome 20 [del(20)(q11.22)] as the sole abnormality (Figure 23-7). Based on the karyotypic change of del(20q), which one of the following interpretations regarding the diagnosis and/or prognosis is correct? B. Deletion 20q as the sole abnormality is associated with MDS and predicts a good prognosis. C. Deletion 20q as the sole abnormality is diagnostic of a specific subtype of AML and predicts a poor prognosis. D. Deletion 20q as the sole abnormality predicts a poor prognosis in MDS. E. Deletion 20q in a complex karyotype predicts a good prognosis in MDS. 14. During a genetic counseling session, a couple report that a third cousin has Down syndrome. There is no other family history of Down syndrome. Which one of the following statements describes the most likely risk for this couple to have a child with Down syndrome? A. Low, because the couple already has six children and none of them have Down syndrome. B. High, as most Down syndrome cases are inherited. C. The same as the age-related risk of the mother. D. High, because the mother of the child with Down syndrome was 22 when the child was born. E. Low, because the cousin is on the father’s side of the family. 15. A woman has an amniocentesis for prenatal chromosome diagnosis because she is a carrier of a Robertsonian translocation. Her karyotype is 45,XX,der(13;21)(q10;q10). Interphase FISH on the uncultured cells from the amniotic fluid sample shows the following signal pattern in 50 nuclei examined: Probe set B: 2 Aqua, 1 Green, 1 Red The probe sets (A and B) used for detection of aneuploidy in this FISH assay are as follows: Locus-specific probe for chromosome 21q22.13 region: Red Locus-specific probe for chromosome 13q14 region: Green Probe set B: Centromeric probe for chromosome 18: Aqua Centromeric probe for the X chromosome: Green Centromeric probe for the Y chromosome: Red This observed signal pattern in the cells from the amniotic fluid sample is most consistent with which one of the following karyotypes? B. 45,XY,der(13;21)(q10;q10). C. 46,XY,der(13;21)(q10;q10),+21. D. 46,XY,der(13;21)(q10;q10),+13. E. 46,XY,der(13;21)(q10;q10),+18. 16. A 63-year-old female patient presented with lymphadenopathy. A submandibular lymph node was excised and a portion submitted for cytogenetic workup. Histologically, the lymph node showed scattered lymphoid follicles and increased numbers of large centroblast-like cells. Immunohistochemical staining was positive for CD20, CD79a, and CD10, and negative for cyclin D1 and MUM1. Cytogenetic analysis (Figure 23-8) showed a complex karyotype that included a translocation between chromosome regions 14q32 and 18q21, resulting in t(14;18). Based on the karyotype observation of t(14;18) and the other chromosome changes, what is the most likely diagnosis of this neoplastic disorder? B. HL. C. Multiple myeloma. D. MALT lymphoma. E. Follicular lymphoma (FL). Figure 23-9 shows a representative FISH image from an analysis performed on cells prepared from amniotic fluid. The image shows the FISH signals from a commercial panel of chromosome enumeration (CEP) for the centromeric regions of chromosomes X, Y, and 18. The X chromosome is represented in green, the Y chromosome in red, and chromosome 18 in aqua. 17a. If all the remaining chromosomes are normal, what is the predicted karyotype based on this hybridization pattern? B. 47,XXY. C. 47,XX,+18. D. 47,XY,+18. E. 46,XY. 17b. What is the correct International System for Human Cytogenetic Nomenclature (ISCN) designation for this image? A. ish(DXZ1x1,DYZ3x1,D18Z1 × 2). B. 46,XX.ish(CEP-X,CEP-Y)x1,(CEP-18) × 2. C. 46. nuc ish(CEP X,Y,18,18). D. nuc ish(DXZ1x1,DYZ3x1,D18Z1 × 2). E. nuc ish(X,Y,18,18). 18. Figure 23-10 shows a representative FISH image from an analysis performed on cells prepared from chorionic villi obtained from a 10-week gestational-aged fetus with anomalies observed by ultrasound. The signals correspond to the following chromosomes as follows: chromosome 13 (red), chromosome 16 (aqua), chromosome 18 (purple), chromosome 21 (green), and chromosome 22 (yellow). Which one of the following is the most likely diagnosis based on the observed hybridization pattern? B. A fetus with tetraploidy. C. A fetus with trisomy 13, trisomy 16, trisomy 18, trisomy 21, and trisomy 22. D. A fetus with malignant transformation. E. Uncertain, because the multiple signals observed are more likely an indication of assay failure. A repeat FISH analysis is indicated. 19. A sample from a patient with a suspected myeloid disorder and with an abnormal eosinophil component was submitted for cytogenetic workup. Karyotype analysis identified inv(16)(p13.1q22) (Figure 23-11, A). FISH analysis using a core-binding factor subunit β (CBFB) break-apart probe showed rearrangement of this gene (Figure 23-11, B, C). Based on the karyotypic change of inv(16) and rearrangement of the CBFB locus by FISH, which one of the following interpretations regarding diagnosis and prognosis is correct? B. Inversion 16 and CBFB gene rearrangement are diagnostic of a specific subtype of AML and predict a good prognosis. C. Inversion 16 and CBFB gene rearrangement are diagnostic of a specific subtype of AML and predict a poor prognosis. D. Inversion 16 and CBFB gene rearrangement are diagnostic of a specific subtype of AML, but have no prognostic value. E. Inversion 16 and CBFB gene rearrangement are diagnostic of ALL. 20. A bone marrow specimen was submitted for cytogenetic workup of a 55-year-old woman who presented with a hypercellular bone marrow with trilineage hematopoiesis, along with a focal interstitial lymphoplasmacytic infiltrate. The patient had a previous history of lymphoma of the small bowel. Immunohistochemical stains showed predominantly CD3-positive T cells and rare CD20-positive B cells. Chromosome analysis (Figure 23-12, A) showed a translocation between chromosome regions 11q21 and 18q21, resulting in a t(11;18) translocation. FISH analysis, using both a MALT1 break-apart probe and a MALT/BIRC3 translocation probe, showed 5% of cells with this rearrangement, thereby confirming the t(11;18) translocation. Based on the karyotype identification of t(11;18) and FISH positivity of MALT1/BRIC3 rearrangement, what is the most likely diagnosis of this lymphoma? B. HL. C. FL. D. MALT lymphoma. E. Splenic marginal zone lymphoma (SMZL). 21. A peripheral blood sample from a 72-year-old female patient with lymphocytosis was submitted for cytogenetic workup for a suspected lymphoma. G-banding karyotype analysis was performed (Figure 23-13), which showed a balanced translocation between chromosomes 11q13 and 14q32, an isochromosome of the long arm of chromosome 17, and loss of chromosome 21. The presence of t(11;14) is a characteristic feature of which one of the following types of lymphoma? B. Mantle cell lymphoma. C. MZL. D. MALT lymphoma. E. Diffuse large B-cell lymphoma. A female child with dysmorphic features, mild mental retardation, a ventricular septal defect, ocular coloboma, preauricular skin tags and pits, and anal atresia has a cytogenetic workup that shows a supernumerary marker chromosome (SMC), 47,XX,+mar (Figure 23-14). Single-nucleotide polymorphism (SNP) microarray analysis is ordered and reveals that the SMC derives from chromosome 22. The extra genomic material appears to be present in a total of four copies, indicating that the SMC is an inversion duplication of the 22p11.2-22q11.21 region, including the TBX1 gene. 22a. The patient’s karyotype and clinical features are most consistent with which one of the following clinical diagnoses? B. Cat-eye syndrome. C. Angelman syndrome. D. Charcot-Marie-Tooth syndrome type 1A. E. Beckwith-Wiedemann syndrome. 22b. Precise identification of the origin of marker chromosomes derived from chromosome 22 using commercial centromeric FISH probes is not possible because of which one of the following reasons? A. Most markers derived from chromosome 22 are mosaic. B. Commercial chromosome 22 probes are not approved by the Food and Drug Administration (FDA). C. Chromosomes 14 and 22 share common α-satellite sequences. D. There are no commercial probes for chromosome 22. E. Most markers derived from chromosome 22 also contain chromosomal sequences from chromosome 18. An amniocentesis specimen of a 24-year-old woman is found to have a de novo supernumerary marker chromosome (SMC). FISH studies indicate that this marker chromosome is derived from chromosome 15, because the probe for the centromere of chromosome 15 hybridized brightly to the marker chromosome. Results of FISH studies using the small nuclear ribonucleoprotein polypeptide N (SNRPN) probe, which hybridizes to the 15q11.2 region, were negative. Additional molecular studies reveal biparental inheritance of the normal chromosome 15 homologues. 23a. Which is the best estimate of the overall risk of an abnormal pregnancy outcome associated with these findings? A. Not significantly increased over the background risk of 3% to 4%. B. High, because chromosome 15 contains imprinted genes. C. Negligible, because there are no imprinted genes on chromosome 15. D. High, because the marker chromosome likely resulted from a rescue of a trisomy 15 fetus. E. Unclear, because further molecular testing is required before accurate risk assessment can be made. 23b. Which chromosomal characteristic is the most likely cause of the clinical features of the majority of cases of Prader-Willi syndrome (PWS)? A. A balanced translocation between the short arms of chromosomes 4 and 5. B. An unbalanced translocation that always involves the proximal long arm of chromosome 22. C. An insertion of extra chromosomal material into the distal long arm region of chromosome 17. D. A duplication of chromosomal material at the proximal region of the long arm of chromosome 7. E. A deletion on the proximal region of the long arm of chromosome 15. 24. Which method can best detect a small deletion on the short arm of chromosome 17 associated with Smith-Magenis syndrome (SMS)? A. A whole chromosome paint probe specific for chromosome 17. B. Spectral karyotyping (SKY) using multicolor chromosome paint probes for all chromosomes. C. A locus-specific FISH probe for the commonly deleted region on chromosome 17. D. A centromere 17–specific FISH probe. E. A subtelomeric probe for the short arm of chromosome 17. 25a. A dysmorphic male child with lissencephaly (i.e., “smooth brain” with pachygyria, incomplete or absent gyration of the cerebrum) has the following karyotype: 46,XY,der(17)t(15;17)(p11.2;p13.3). Which one of the following statements best describes the child’s karyotype? A. The karyotype shows a balanced translocation between the short arms of chromosomes 15 and 17. B. The karyotype shows an unbalanced translocation between the short arms of chromosomes 15 and 17, which results in partial trisomy of 15p and partial monosomy of 17p. C. The karyotype shows an unbalanced translocation between the short arms of chromosomes 15 and 17, which results in partial monosomy of 15p and partial trisomy of 17p. D. The karyotype shows an unbalanced derivative chromosome 17 resulting from a translocated insertion of short arm material from chromosome 15p at band p13.3 into 17p11.2. E. The karyotype shows an unbalanced derivative chromosome 17 resulting from a translocated insertion of short arm material from chromosome 15p at band p11.2 into 17p13.3. 25b. Which clinical diagnosis is most consistent with this patient’s karyotype and clinical features? B. SMS. C. Cri-du-chat syndrome. D. DiGeorge syndrome. E. Miller-Dieker syndrome. 25c. Which statement best describes the phenotype associated with the following karyotype: 46,XY,der(17)t(15;17)(p11.2;p13)? A. A sequence-based mutation because the karyotype is balanced. B. The missing genetic material from the short arm of chromosome 15 and the extra genetic material from the short arm of chromosome 17. C. The extra genetic material from the short arm of chromosome 15 and the missing genetic material from the short arm of chromosome 17. D. Only the missing genetic material from the short arm of chromosome 17. E. Only the extra genetic material from the short arm of chromosome 15. 26. A bone marrow specimen from an 89-year-old male patient with paraproteinemia was submitted to the cytogenetics laboratory. The findings in the hematopathology laboratory identified CD5-positive cells and κ-restriction of the neoplastic cells. Chromosome analysis performed on the bone marrow showed a complex karyotype with both copies of chromosome 14 rearranged at the 14q32 band, as shown in Figure 23-15, A. The karyotype was described as follows: B. MZL. C. Multiple myeloma, possibly with good prognosis. D. Multiple myeloma, possibly with poor prognosis. E. MALT lymphoma. 27. A 75-year-old male patient presented with an intestinal obstruction and perforation secondary to the presence of a jejunal mass, which was submitted for cytogenetic workup. Microscopic analysis of the jejunal mass showed a dense and diffuse proliferation of large lymphoid cells spanning the entire thickness of the small bowel wall. Immunohistochemistry showed that these cells were CD20+, CD79a+, CD10+, Bcl6+, CD5–, and MUM1–. The proliferation rate was approximately 70% as determined by Ki-67 immunostaining, and Epstein-Barr virus–encoded small RNA (EBER) was negative by in situ hybridization. Flow cytometric analysis, after preferential gating of CD45 bright cells, showed a κ-restricted B-cell population with the phenotype of CD19+, CD20+, CD10+, cytoplasmic CD79a+, CD34–, CD5–, CD23–, HLA-DR+, CD38+, CD52+, CD43–/+, sIgM+. Chromosome analysis (Figure 23-16, A) showed a complex karyotype, including a translocation between chromosome regions 8q24 and 2p11.2, resulting in t(2;8), several gains of chromosomes, losses of chromosomes, and deletions of chromosomes. Based on the morphologic, immunohistochemical, flow cytometric, and cytogenetic observation of the t(2;8) translocation associated with a complex karyotype, what is the most likely diagnosis of this neoplasm? A. High-grade B-cell non-Hodgkin lymphoma (B-NHL). B. MZL. C. Multiple myeloma. D. MALT lymphoma. E. HL. 28. Which one of the following chromosome abnormalities is associated with a poor prognosis in patients with MDS? B. 47,XY,+8. C. 46,XY,del(20)(q11.2q13.3). D. 45,X,-Y. E. 46,XY,del(7)(q11.2q36). 29. Deletions of chromosomes 1p and 19q are of diagnostic and prognostic significance in which one of the following types of tumors of the nervous system? B. Medulloblastomas. C. Meningiomas. D. Oligodendrogliomas. E. Ependymomas. 30. A right kidney mass obtained after partial nephrectomy from a 55-year-old male patient was submitted for cytogenetic workup. Karyotype analysis showed gains of chromosomes 3, 7, 12, 16, and 17, and loss of the Y chromosome (Figure 23-17). Based on the karyotypic gains of these chromosomes, what is the most likely diagnosis? A. Chromophobe-type renal cell carcinoma (RCC). B. Renal oncocytoma. C. Papillary RCC. D. Conventional clear cell RCC. E. Renal angiomyolipoma. 31. A 16-year-old girl presented with a right shoulder mass, which was submitted for cytogenetic workup. The findings in surgical pathology identified tumor cells that were positive for CD99, beta-catenin, vimentin, and synaptophysin. Chromosome analysis performed after short-term cell culture showed a highly abnormal karyotype, as shown in Figure 23-18, A. Based on the breakpoint at 22q12, FISH analysis was performed using an EWSR1 dual-color break-apart probe. Based on the karyotype and FISH analysis, what is the most likely diagnosis of this tumor? A. Embryonal rhabdomyosarcoma. B. Primitive neuroectoderal tumor. C. Giant cell tumor of bone. D. Lipoma. E. Alveolar rhabdomyosarcoma. 32. A bone marrow specimen from a 10-month-old female infant with persistent fever, but no significant past medical history, and suspicion of acute leukemia, was submitted for chromosome analysis. Histological examination of a bone marrow biopsy revealed complete replacement of normal hematopoietic elements with atypical, small- to medium-sized lymphocytes with irregular nuclear contours, dispersed chromatin, indistinct nucleoli, and little to no cytoplasm. Numerous mitotic figures were seen. Flow cytometric analysis of the marrow after exclusion of the CD45-negative to dim gate, showed a phenotype of CD19+, CD79a+, CD22+, CD10+, CD20–, CD34+ (partial), CD117–, HLA-DR+, CD43+, CD38+, CD52+, and TdT+. Chromosome analysis showed a gain of specific chromosomes (Figure 23-19). This karyotype is described as 52,XX,+6,+8,+16,+19,+21,+22[15]/46,XX[5]. Based on this karyotype, combined with the morphological and flow cytometric findings, which statement best describes the most likely diagnosis and prognosis in this case? A. Chronic myeloid leukemia (CML), predicting a poor prognosis. B. MDS, predicting a good prognosis. C. Pre-B-cell ALL (pre-B ALL), predicting a poor prognosis. D. Pre-B ALL, predicting a good prognosis. E. AML, predicting a good prognosis. 33. A sample from an 11-year-old boy with a history of easy bruising, pancytopenia, and blasts on peripheral smear was submitted for cytogenetic analysis. The karyotype analysis identified t(15;17)(q22;q21) (Figure 23-20, A); FISH analysis using promyelocytic leukemia (PML) and retinoic acid receptor alpha (RARA) gene probes showed rearrangement between these genes (Figure 23-20, B). Based on the karyotypic identification of 46,XY,t(15;17)(q22;q21) and the FISH identification of rearrangement between the PML and RARA loci, which statement is correct? A. The t(15;17)(q22;q21) translocation is commonly seen in MDS. B. The t(15;17)(q22;q21) translocation is diagnostic of CML. C. The t(15;17)(q22;q21) translocation is diagnostic of APL but does not predict treatment outcome. D. The chromosomal translocation (15;17)(q22;q21) is not associated with acute myelocytic leukemia. E. The t(15;17)(q22;q21) translocation is diagnostic of APL and predicts a favorable treatment outcome. 34. A portion of a kidney mass obtained after radical nephrectomy from a 73-year-old male patient was submitted for cytogenetic workup. Karyotype analysis showed the loss of chromosomes 1, 2, 5, 6, 10, 13, 17, 21, and the Y chromosome (Figure 23-21). In addition, a t(3;8)(p21.3;p12) and trisomy 8 were identified. Based on the karyotypic loss of chromosomes, what is the most likely diagnosis? B. Renal oncocytoma. C. Papillary RCC. D. Conventional clear cell RCC. E. Renal angiomyolipoma. 35. Deletion of the short arm of chromosome 3 is a characteristic finding in which one of the following types of RCC? B. Papillary RCC. C. Chromophobe RCC. D. Mucinous tubular and spindle cell carcinoma. E. Oncocytoma. 36. What is the correct ISCN 2009 nomenclature for a female carrier of a balanced Robertsonian translocation involving chromosomes 13 and 21? B. 45,XX,der(13;21)(q10;q10). C. 46,XX,t(13;21)(p10;q10). D. 46,XX,der(13),t(13;21)(p10;p10). E. 46,XX,der(13;21)(q10;p10). 37. A solid tumor from a 25-year-old patient shows a translocation (X;18)(p11.2;q11.2). This is a characteristic finding in which setting? B. Desmoplastic small round cell tumor. C. Embryonal rhabdomyosarcoma. D. Alveolar rhabdomyosarcoma. E. Myxoid liposarcoma. 38. A lung nodule resection of a metastatic tumor from a 25-year-old female patient was submitted for cytogenetic workup of a soft tissue neoplasm. The findings in surgical pathology identified tumor cells composed of intersecting highly cellular bundles of spindle shaped cells. Immunohistochemical stains were positive for EMA, ESO-1, and PDGFR-β and negative for HMB-45, S100, cytokeratin, HHF35F, and c-kit. Chromosome analysis performed after short-term cell culture showed an abnormal karyotype (Figure 23-22, A). Based on the breakpoints at Xp11.2 and 18q11.2, including other abnormalities, the karyotype was described as 46,X,t(X;18)(p11.2;q11.2),der(21;22)(q10;q10)[12]/46,XX[8]. Based on the t(X;18) translocation identified by karyotype analysis, which one of the following is the most likely diagnosis? A. Embryonal rhabdomyosarcoma. B. Alveolar rhabdomyosarcoma. C. Primitive neuroectoderal tumor. D. Synovial sarcoma. E. Myxoid liposarcoma. 39. A peripheral blood sample from a 52-year-old male patient was submitted for cytogenetic workup to rule out lymphoma. Flow cytometric analysis, after preferential gating for CD45 bright cells, showed a predominant population of small CD4+ T cells with the following phenotype: CD3+, CD4+, CD8–, CD2+, CD7+, CD5+, TCR-αβ+, CD43+, CD38+, CD25–, and CD52+. The peripheral blood cells were negative for B-cell markers, myeloid markers, and immaturity markers. Chromosome analysis (Figure 23-23, A) showed a simple karyotype consisting of a paracentric inversion of the long arm of chromosome 14, involving regions between q11.2 and q32, resulting in inv(14)(q11.2q32) (Figure 23-23, B) and a deletion on the short arm of chromosome 9p22 [del(9)(p22)]. Based on the flow cytometric findings and the cytogenetic observation of inv(14)(q11.2q32), what is the most likely diagnosis of this lymphoma? B. ALCL. C. Multiple myeloma. D. T-PLL. E. HL. 40. Which of the following karyotypes is most likely to be found in a newborn female with multiple congenital abnormalities and dysmorphic features? B. 46,XX,t(7;18)(q21;q21). C. 47,XX,+16. D. 47,XXX. E. 46,XX,inv(9)(p12q13). 41. Which woman is at greatest risk of having a child with trisomy 21 (Down syndrome)? A. A 30-year-old woman with a 46,XX karyotype. B. A woman with the karyotype 45,XX,t(21;21)(q10;q10). C. A woman with no family history of Down syndrome. D. A woman with the karyotype 45,XX,t(14;21)(q10;q10). E. A 35-year-old woman who had a previous child with trisomy 21. Major points of discussion ■ This method is used to detect any unbalanced chromosome abnormalities. Unbalanced abnormalities include gains and losses of (1) entire chromosomes (aneuploidy), (2) small individual segments of DNA that cannot be detected by standard chromosome analysis, (3) extra SMCs, and (4) ring chromosomes. This method is used in both constitutional and cancer cytogenetics. ■ Depending on the resolution of the chip (the number of probes or oligonucleotides and the spacing between the probes or oligos across the genome), small deletions or duplications can be identified with detailed information about the genomic region involved: size, base positions, number of genes, and genomic content. ■ Truly balanced chromosome rearrangements, such as inversions and balanced translocations, cannot be detected by this method. ■ This method has revealed previously unappreciated normal variation in the number of copies of DNA segments known as copy number variants.60 2. A. Burkitt lymphoma (BL) with a poor prognosis. Major points of discussion ■ The chromosome changes in B-cell ALL play important roles with regard to diagnostic classification and prognosis prediction. ■ A major focus concerning the pathogenetic role of chromosomal abnormalities in ALL has centered on the structural changes typical of this disease. The most commonly seen chromosome translocations in ALL are t(12;21)(p13;q22), translocations involving 11q23 with various different chromosomes, t(9;22)(q34;q11.2), and t(1;19)(q23;p13.3). ■ The MLL gene at 11q23 is rearranged with multiple chromosome regions in ALL. The most frequent reciprocal translocation are t(4;11)(q21;q23), t(9;11)(p21;q23), t(6:11)(q27;q23), and t(11;19)(q23;p13). Among the MLL rearranged translocations in ALL, t(4;11) is the most common abnormality. ■ The t(4;11)(q21;q23) translocation leads to a fusion between the MLL gene and AF4 (AFF1) gene at 4q21, thereby generating an MLL-AFF1 fusion transcript. This translocation is strongly associated with ALL in infants and is also seen in a small proportion of acute myelocytic leukemia cases. ■ In general, leukemias with the t(4;11)(q21;q23) translocation exhibit a poor prognosis. In particular, infant ALL patients carrying t(4;11) have a high relapse rate and a poor outcome.24,51 3. A. Natural killer (NK)-cell leukemia and lymphoma. Major points of discussion ■ The most common chromosome translocation in ALCL, in approximately 80% of cases, is a balanced translocation between chromosome regions 2p23 and 5q35. ■ The molecular consequence of the t(2;5)(p23;q35) translocation is the fusion of the nucleophosmin (NPM) gene at 5q35 with the ALK gene at 2p23, resulting in the formation of a chimeric protein with constitutive kinase activity. ■ Variant translocations involving ALK with other partner genes on chromosomes 1, 2, 3, 17, 19, 22, and X have been reported. Examples include t(1;2)(q21-q25;p23) fusing ATM with TPM3, t(2;3)(p23;q12) leading to a TFG-ALK fusion, inv(2)(p23q35) leading to a ATIC-ALK fusion, and t(X;2)(q11;p23) leading to MSN-ALK fusion. All of the translocations result in up-regulation of ALK expression. ■ The ALK gene encodes a receptor tyrosine kinase, which is normally silent in lymphoid-lineage cells. ■ When ALK is rearranged in ALCL cases, it predicts a favorable prognosis, irrespective of the translocation partner, with a 5-year overall survival rate of approximately 80%. ■ ALK-negative ALCL exists, which is considered as a different biologic entity from ALK-positive ALCL, and has a poor prognosis. ■ The ALK gene is also rearranged in non–small cell lung cancer due to paracentric inversion of 2p, resulting in an ALK/EML4 fusion gene. ALK is also amplified in neuroblastoma, and mutations of ALK predispose patients to neuroblastoma.2,34,47 4. A. BL, predicting a poor prognosis. Major points of discussion ■ The chromosome changes in B-cell ALL play an important role regarding diagnostic classification and prognosis prediction. ■ Ploidy groups in ALL represent well-established cytogenetic entities comprising low hyperdiploidy (46 to 49 chromosomes), high hyperdiploidy (51 to 65 chromosomes), near-triploidy (66 to 79 chromosomes), near-tetraploidy (84 to 100 chromosomes), hypodiploidy (45 chromosomes), low hypodiploidy (31 to 39 chromosomes), and near-haploidy (25 to 29 chromosomes). ■ Each ploidy group predicts chemotherapy responses in ALL. Overall, hyperdiploid ALL groups respond well to standard chemotherapy compared with patients with hypodiploid and near-haploid karyotytes. ■ Children with ALL with hypodiploidy karyotypes have a progressively worse outcome with decreasing chromosome numbers, even after treatment with intensive protocols. ■ The near-haploid karyotype is rare, and this unique ALL patient group presents more frequently in children than in adults. ■ A near-haploid karyotype is defined as having 25 to 29 chromosomes, and the majority of such cases have 26 chromosomes. This chromosome loss is not random. There is a preferential retention of disomy of chromosomes 21, X, Y, 14, and 18, in this order of frequency. Structural chromosome abnormalities are rarely seen in the near-haploid group. A near-haploid clone can also be present along with a hyperdiploid karyotype, as a result of duplication of the near-haploid karyotype. Distinguishing a hyperdiploidy clone resulting from duplication of near-haploid cells from a true hyperdiploid karyotype is extremely important, given the very different prognostic significance of these two patient groups. ■ Childhood ALL patients with a near-haploid karyotype generally have a short complete remission and a dismal prognosis.10,24,29 5. A. 46,XY,(8;21)(q22;q22). Major points of discussion ■ The salient pathologic features of AML include the excessive accumulation of immature myeloid blasts in the bone marrow. This maturation block, a characteristic of acute leukemias, prevents normal hematopoiesis and leads to a lack of differentiated granulocytes, monocytes, thrombocytes, and erythrocytes. ■ The World Health Organization (WHO) classification of AML includes morphologic, immunophenotypic, genetic, and clinical features. Four categories are delineated: (1) AML with recurrent translocations, which include (8;21), (15;17), and 11q23 translocations, and inversion(16)/translocation (16;16); (2) AML with multilineage dysplasia; (3) therapy-related AML (tAML) and myelodysplatic syndromes (tMDS); and (4) AML not otherwise categorized. ■ The classic t(15;17) translocation is a characteristic finding in APL and results in a PML/RARA gene fusion. This is an important cytogenetic diagnosis because of its responsiveness to all trans-retinoic acid (ATRA) therapy, which promotes differentiation of the leukemic cells. ■ About 20% to 30% of pediatric and 40% to 50% of adult AML patients do not have any cytogenetically identified chromosome abnormality. AML with normal karyotype has been shown to have various genetic abnormalities, which include submicroscopic deletions, duplications, and uniparental disomies (which can be detected by aCGH or SNP array); thus these cases are not normal at the genomic level. Mutation analysis of genes such as FLT3, NPM1, and CEBPA is also used in clinical practice. 6. A. BL. Major points of discussion ■ The t(8;14)(q24;q32) translocation is a diagnostic feature of BL, which is seen in 75% to 85% of all such cases. This translocation results in juxtaposition of the IGH enhancer to the MYC oncogene, resulting in the latter’s deregulated expression. ■ Variant translocations, such as t(8;22)(q24;q11.2) and t(2;8)(p12;q24), which involve rearrangement of the immunoglobulin light chain genes, IGL and IGK, respectively, with the MYC gene are seen in the remaining 15% to 25% of cases of BL. ■ None of the three types of chromosomal translocations described above are completely specific for BL. These three types of translocations can be seen rarely in almost all histologic subtypes of B-cell lymphomas. ■ Secondary chromosome abnormalities are seen in approximately 60% of cases of BL and are sometimes indicators of disease progression. The most frequent structural aberration is a 1q duplication. Other common secondary changes in BL are trisomies 7 and 12.6,7,42 7. A. BL. Major points of discussion ■ The t(8;14)(q24;q32) translocation is a diagnostic feature of individuals with BL, which is seen in 75% to 85% of all cases. ■ Variant translocations, such as t(8;22)(q24;q11.2) and t(2;8)(p12;q24), which involve rearrangement of the MYC gene with the immunoglobulin light chain IGL and IGK genes, respectively, are seen in 15% to 25% of cases of BL. The t(8;22) translocation results in juxtaposition of the IGL enhancer with the MYC oncogene and thereby deregulates its expression. ■ The t(8;22)(q24;q11.2) translocation is not completely specific for BL. It can also be seen, albeit rarely, in almost all histologic subtypes of B-cell lymphomas and in ALL with L3 morphology. ■ The t(8;22)(q24;q11.2) translocation occurs in both endemic and sporadic forms of BL. ■ There is a molecular heterogeneity in terms of the breakpoints in the MYC gene between the t(8;14), t(8;22), and t(2;8) translocations. The MYC breakpoints in t(2;8) and t(8;22) are telomeric to MYC; in t(8;14), the breakpoint in MYC is centromeric.6,42,61 8. A. The karyotype (Figure 23-6, A) predicts a good prognosis. Major points of discussion ■ Common recurrent chromosome aberrations in CLL are trisomy 12, deletion 13q14.3 (a likely target of micro RNA genes miR-16-1 and miR-15a), deletion 11q22 (a likely target of the ATM gene), and deletion 17p13 (a likely target of the TP53 gene). ■ Specific cytogenetic abnormalities are known to be important independent predictors of prognosis. For example, deletion 13q or monosomy 13 predict a good prognosis. TP53 (17p13) and ATM (11q22) deletions individually, or in combination, predict a poor response to treatment.3,56,59 9. A. 10% to 15%. Major points of discussion ■ The greater the size of the chromosomal aberration (imbalance), the earlier the fetus is expected to miscarry. ■ Between 10% and 15% of all recognized pregnancies end in a spontaneous abortion; of these, approximately 65% to 70% are associated with chromosome abnormalities. ■ All autosomal monosomies are lethal and most trisomies lead to early fetal loss. ■ Aneuploidy accounts for the largest proportion (> 80%) of abnormalities observed in products of conception. ■ A 46,XX result may, in some cases, reflect the maternal karyotype as a consequence of maternal cell contamination. This is largely why gender ratios from published studies are skewed to a higher proportion of female karyotypes. The use of SNP microarray analysis for cytogenomic evaluation of miscarriages offers the ability to detect maternal cell contamination as well as provide a higher-resolution analysis for genomic imbalance.21,40,43 10. A. Their specific bar code, the size of the centromere, and the amount of heterochromatin. Major points of discussion ■ When the position of the centromere is located off-center, such that there is a distinct difference in the sizes of the chromosomal arms, the chromosome is described as a “submetacentric” chromosome. The submetacentric chromosomes are 2, 4, 5, 6, 7, 8, 9, 10, 11, 12, 17, 18, and X. ■ When the position of the centromere is located at the tip of the short arm, such that the long arm comprises almost the entire chromosome, the chromosome is described as an “acrocentric” chromosome. The acrocentric chromosomes typically refer to chromosomes 13, 14, 15, 21, and 22. However, the Y chromosome can also be considered an acrocentric chromosome. ■ The chromosomes that have large heterochromatic blocks are 1, 9, 16, and Y. These blocks are composed of large arrays of tandemly repeated DNA elements. Evolutionary processes acting on these DNA blocks played a role in expansion of arrays within a particular chromosomal location. As a result, the size of these blocks became polymorphic and size variations between individuals are routinely observed. The size of the heterochromatic blocks tends to be inherited in a stable manner and, therefore, is identical to the parent from which the individual has inherited the chromosome. ■ GTG (G bands produced with trypsin using Giemsa) banding is the most widely used staining method for producing the characteristic G bands that are used to identify each chromosome uniquely. ■ C-banding stains constitutive heterochromatin around the centromeres and the large heterochromatic regions found on chromosomes 1, 9, 16, and Y. CBG (C bands produced with barium hydroxide using Giemsa) banding is a common technique for producing C bands. ■ Telomeres serve to stabilize chromosome ends and are composed of repeating units of the (TTAGG)n DNA sequence.4,39,54 11. A. l-Isoleucine. Major points of discussion ■ Cell culture medium should be constantly monitored for sterility. Most commercial media contain a pH indicator, phenol red, which changes color when contamination is present. ■ Antibiotics and antifungal agents are typically added to control growth of bacterial/fungal contaminants. ■ Serum (typically fetal bovine serum) contains growth factors required for cell viability and proliferation. ■ Typical cytogenetic growth media contain buffered solution with phenol red, sugars, amino acids, ions, and supplements (serum, antibiotics, antifungal agents, l-glutamine).4,35 12. A. This result indicates a sample mix-up in the laboratory with the POC being triploid and the normal female cells coming from another specimen in the laboratory. Major points of discussion ■ Triploidy could be diandry (two copies of the paternal genome) or digyny (two copies of the maternal genome). ■ Diandry usually results from dispermy; that is, two sperm fertilizing the oocyte. In rare instances, diandry results from a complete nondisjunction in the sperm that fertilizes the oocyte. Digyny results from complete nondisjunction in the first or second meiotic division of the oocyte. ■ Most diandric triploid fetuses spontaneously abort in the first trimester. The very few that survive the second trimester are partial hydatidifrom moles. Digynic triploids spontaneously abort early in the first trimester. ■ There is one case report of a digynic triploid (69,XXX) coexisting with a normal twin (46,XY); however, this is very rare. ■ Intrauterine survival into the third trimester is possible in cases of fetal-placental discordance; that is, the placenta is diploid and the fetus is triploid. However, this is almost invariably associated with perinatal death. Rare instances of survival for more than 1 month are known.20,23,31 13. A. Deletion 20q as the sole abnormality is not associated with myeloid neoplasms, such as MDS or AML. Major points of discussion ■ Deletion 20q as an isolated abnormality is seen in 5% to 7% of cases of isolated MDS and treatment-related MDS. Isolated 20q deletion is associated with a favorable outcome in MDS according to the IPSS. When deletion of 20q is the sole abnormality in MDS patients, it predicts a low risk for progression to AML and these patients have a prolonged survival. ■ When MDS patients carry del(20q) as a part of a more complex karyotype, they are at risk for a poor outcome. ■ Deletion 20q is also found in 1% to 2% of all cases of AML, and very few AML patients carry this as the sole chromosome change. However, deletion 20q is not specific for any particular FAB subtype of AML. In addition, the prognostic impact of del(20q) as a single chromosomal abnormality in AML is unclear. ■ Deletion 20q is frequently associated with other chromosome changes in MDS and AML patients, such as del(5q), trisomy 8, and trisomy 21.9,25,27 14. A. Low, because the couple already has six children and none of them have Down syndrome. Major points of discussion ■ Down syndrome is recognized as the most common single known cause of intellectual disability. ■ Down syndrome has the highest incidence at birth of any chromosome abnormality. ■ The majority of Down syndrome is due to nondisjunction leading to trisomy of chromosome 21. ■ The majority (~ 90%) of nondisjunction events reflect maternal meiotic errors. ■ Approximately 75% of maternal meiotic errors occur at meiosis I and the remainder at meiosis II.21,38,54 15. A. 46,XY. Probe set B: 2 aqua, 1 green, 1 red B. 45,XY,der(13;21)(q10;q10). Probe set B: 2 aqua, 1 green, 1 red C. 46,XY,der(13;21)(q10;q10),+21. Probe set B: 2 aqua, 1 green, 1 red D. 46,XY,der(13;21)(q10;q10),+13. Probe set B: 3 aqua, 1 green, 1 red Major points of discussion ■ Interphase FISH using this probe set is routinely used for rapid detection of aneuploidies in prenatal samples. However, this probe set provides limited information. ■ This probe set will not detect aneuploidies of chromosomes other than those of 13, 21, 18, X, and Y. Structural abnormalities can also not be detected because the probes are specific for either the centromeric regions or a particular locus on the chromosome. ■ A full chromosome study is required to distinguish structural rearrangements; for example, the karyotypes described in choices A and B will have the same signal pattern. However, from this signal pattern, it cannot be discerned whether the fetus has a normal karyotype (46,XY) or is a carrier of a Robertsonian translocation 45,XY,der(13;21)(q10;q10). ■ Karyotyping from cultured cells is required for a full chromosome study.60 16. A. BL. Major points of discussion ■ The t(14;18)(q32;q21) translocation is a diagnostic feature of FL. ■ In addition to t(14;18), other secondary chromosome alterations, such as deletions on 6q, 17p, 1p36, trisomy 7, and translocations involving 3q27 (the BCL6 gene), are also seen in FL. ■ Secondary chromosome abnormalities are sometimes indicators of disease progression, transformation to diffuse large B-cell lymphoma, or prognosis. For example, trisomy 7 and 6q and 17p deletions are associated with morphologic progression. Tumors with the 3q27 rearrangement can progress to FL. The 6q and 1p36 deletions are associated with an unfavorable outcome.3,56,59 17a. A. 46,XX. 17b. A. ish(DXZ1x1,DYZ3x1,D18Z1 × 2). Major points of discussion ■ The abbreviation “nuc ish” is followed, in parentheses, by the locus designation, multiplication sign (×), and the number of signals seen. ■ If probes for two or more loci are used in the same hybridization, they follow one another in a single set of parentheses, separated by a comma (,) and a multiplication sign (×) outside the parentheses if the number of signals for each probe are the same, and inside the parentheses if the number of hybridization signals varies. ■ All interphase FISH studies performed on cancer specimens should indicate the number of cells scored in square brackets [ ] following the hybridization results. ■ Results from prenatal FISH studies on direct chorionic villi are typically available within 24 to 48 hours.54 18. A. A fetus with triploidy. Major points of discussion ■ Diandry is the term used to describe triploidy when the extra chromosome set derives from the father. ■ The majority of diandry triploidy is caused by the fertilization of a single ovum by two sperm. ■ Digyny is the term used to describe triploidy when the extra chromosome set derives from the mother. ■ Digyny is more commonly caused by the fertilization of a diploid ovum by a single haploid sperm. ■ The great majority of triploid conceptions spontaneously abort during the first, or early second, trimester. 19. A. Inversion 16 and CBFB gene rearrangement are NOT diagnostic of any subtype of acute myeloblastic leukemia (AML). Major points of discussion ■ The chromosome changes t(8;21)(q22;q22), t(15;17)(q22;q12-q21), and inversion (16)(p13q22) are diagnostic of specific subtypes of AML and serve as prognostic indicators of treatment outcome. ■ Certain chromosome changes in AML serve as indicators of prognosis. For example, 11q123 translocations with different partner chromosomes predict a generally poor outcome.3,32,59 20. A. BL. Major points of discussion ■ Three major and mutually exclusive recurrent chromosome translocations that target activation of the NFKB pathway are associated with MALT lymphomas. The t(11;18)(q21;q21) translocation is seen as a sole abnormality in approximately 50% of MALT lymphomas associated with the stomach and lung. This translocation results in formation of a BIRC3–MALT1 fusion gene. ■ The second most frequently detected translocation is t(14;18)(q32;q21), which is present in approximately 11% of MALT lymphomas. This translocation is more frequently associated with MALT lymphomas in the liver and ocular adnexa. This translocation juxtaposes the MALT1 gene next to the IGH locus at 14q32. This translocation is cytogenetically identical to the t(14;18)(q32;q21) translocation seen in follicle center cell origin lymphomas; it can only be differentiated by using molecular methods (e.g., FISH and polymerase chain reaction [PCR]). The third translocation, t(1;4)(p22;q32), is detected in less than 2% of MALT lymphomas. This translocation juxtaposes the BCL10 gene at 1q22 next to the IGH locus at 14q32. ■ Translocation-negative MALT lymphomas are associated with chromosomal changes leading to partial trisomy of the long arms of chromosomes 3 and 18. ■ MALT lymphomas have an indolent course and are sensitive to radiation therapy and antibiotic therapy for H. pylori. Tumors harboring the t(11;18) translocation have a lower probability response to H. pylori antibiotic therapy. Occasionally, MALT lymphomas may transform into high-grade, diffuse large B-cell lymphomas.1,19,41 21. A. BL. Major points of discussion ■ Mantle cell lymphoma has a strong male predominance and is considered to be a low-grade B-cell non-HL with a progressive clinical course. ■ The t(11;14)(q13;q32) translocation is the cytogenetic hallmark of mantle cell lymphoma and is considered as a primary genetic event in its genesis. At the molecular level, this translocation juxtaposes the cyclin D1 gene at 11q23 next to the IHC locus at 14q32, resulting in overexpression of the cyclin D1 gene. ■ Other secondary chromosome abnormalities, such as monosomy 13/deletion 13q, trisomy 12, gain of chromosome 3q, and deletion of 9q can often be seen in the blastoid form of mantle cell lymphoma. ■ Secondary chromosome abnormalities are associated with a poor prognosis in mantle cell lymphoma.3,56,59 22a. A. DiGeorge syndrome. 22b. A. Most markers derived from chromosome 22 are mosaic. Major points of discussion ■ The phenotypic consequences of having an SMC are highly variable, ranging from benign to severe. The risk of an adverse phenotypic outcome is correlated with the gene content of the SMC: The larger the gene content, the more likely is the adverse outcome. ■ SMCs occur in approximately 1:3333 consecutive newborns. Their frequency in the mentally retarded population is 10 times higher, occurring in approximately 1:305 cases. ■ Approximately 80% of SMCs derive from the acrocentric chromosomes (13, 14, 15, 21, and 21) and 50% of these come from chromosome 15. ■ Approximately 20% of SMCs are inherited from a normal parent with no increased risk of an adverse outcome expected. ■ FISH using a combination of centromere, locus-specific, or chromosome-specific probes has been used to resolve the chromosomal origin of SMCs. Currently, microarray analysis is the preferred method for identifying the chromosomal origin of SMCs because it elucidates not only the chromosomal origin but also the gene content.13,16,39,48,55 23a. A. Not significantly increased over the background risk of 3% to 4%. 23b. A. A balanced translocation between the short arms of chromosomes 4 and 5. Major points of discussion ■ PWS is a genomic imprinting disorder characterized by the following: • Hypotonia and feeding difficulties in early infancy • Excessive eating in childhood, gradual morbid obesity • Delayed motor milestones and language development • Cognitive impairment • Temper tantrums • Hypogonadism, short stature, strabismus, and scoliosis ■ The critical genomic region implicated in PWS is the proximal region of the long arm of chromosome 15, just below the centromere (15q11.2-q13). ■ The critical genomic region implicated in PWS contains imprinted genes, which means that expression of that gene occurs in a gender-specific manner; that is, expression only occurs from one parent’s allele. The parental allele that is not expressed (i.e., is silent) is referred to as “imprinted.” Thus, a maternally imprinted gene will not be expressed from the maternally inherited chromosome but will only be expressed from the paternally inherited chromosome. ■ Multiple genes in the PWS genomic region are maternally imprinted; thus, expression of those genes occurs from the paternally inherited chromosome. A deletion of this region on the paternally inherited chromosome results in PWS, because expression of these imprinted genes no longer occurs. The deletion is typically interstitial and is usually 5 to 6 Mb in size. ■ Maternal uniparental disomy of chromosome 15 (i.e., both copies of chromosome 15 are derived from the mother; none from the father) effectively results in the same clinical consequence (PWS) because the genes that need to be expressed are missing (i.e., no paternal contribution). ■ A minor number of PWS cases (< 3%) can be caused by imprinting defects that result from epigenetic causes that demonstrate a maternal-only DNA methylation pattern despite the presence of both parental alleles (i.e., biparental inheritance). Approximately 15% of individuals with imprinting defects have a very small deletion (7.5 to 100 kb) in the PWS imprinting center region located at the 5′ end of the SNRPN gene and promoter. ■ Approximately 65% to 75% of PWS cases are due to a deletion of the critical genomic region on the paternally inherited chromosome at 15q11.2. ■ Approximately 20% to 30% of PWS cases are due to maternal uniparental disomy of chromosome 15. ■ Approximately 1% to 3% of PWS cases are due to imprinting defects. ■ Deletions of the same critical genomic on the maternally inherited chromosome at 15q11.2 or paternal uniparental disomy of chromosome 15 lead to a completely different disorder: Angelman syndrome. ■ Angelman syndrome is characterized by the following: • Absent or limited speech • Gait ataxia • Happy demeanor with frequent inappropriate laughter, smiling, and excitability • Microcephaly and seizures ■ Angelman syndrome may also be caused by defects/mutations in the imprinting center or by mutations in the UBE3A gene.11,36,37 24. A. A whole chromosome paint probe specific for chromosome 17. Major points of discussion ■ All deletions associated with SMS include a deletion of the RAI1 gene located on chromosome 17p11.2. This syndrome is characterized by distinctive facial features that progress with age, developmental delay, cognitive impairment, and behavioral abnormalities. Infants have feeding difficulties, failure to thrive, hypotonia, hyporeflexia, prolonged napping, and generalized lethargy. The majority of these individuals have a mild-to-moderate range of intellectual disability. ■ Approximately 90% of the cases with deletion of chromosome 17p11.2 can be detected using a locus-specific probe for the RAI1 gene. Sequence analysis is used in 5% to 10% of the suspected SMS patients when FISH testing is negative for the 17p11.2 deletion. ■ Chromosome paint probes are fluorescently labeled libraries of DNA sequences derived from flow-sorted chromosomes and SKY is a FISH method that allows visualization of all the human chromosomes, each in a different fluorescent color. ■ Recently, aCGH or CMA has become the diagnostic test of choice for detecting microdeletion syndromes.57,60 25a. A. The karyotype shows a balanced translocation between the short arms of chromosomes 15 and 17. 25b. A. Sotos syndrome. 25c. A. A sequence-based mutation because the karyotype is balanced. Major points of discussion ■ Approximately 80% of individuals with Miller-Dieker syndrome have a de novo deletion involving 17p13.3. ■ In approximately 12% to 20% of Miller-Dieker syndrome cases, the deletion at 17p13.3 is due to a familial balanced chromosome rearrangement. ■ The deletion at 17p13.3 is cytogenetically visible in approximately 70% of patients with Miller-Dieker syndrome. Some cases with isolated lissencephaly do not show visible cytogenetic deletions and are more likely to have mutations of the LIS1 gene or tiny deletions restricted to the LIS1 gene. ■ Additional molecular cytogenetic testing should be pursued for those patients with clinical features of Miller-Dieker syndrome that show a normal karyotype. Additional testing can be performed by FISH using a probe that maps to the LIS1 gene region or by DNA microarray analysis.16,17,21,33,48 26. A. BL. Major points of discussion ■ Chromosome abnormalities are found in only approximately 30% of the cases by conventional karyotypic analysis due to the low proliferative index exhibited by multiple myeloma. ■ Karyotypes are generally complex, involving both chromosome number and structure. The most common chromosomes that involve changes in chromosome number are monosomy 13 and trisomies of chromosomes 3, 5, 7, 9, 11, 15, 19, and 21. Structural chromosome abnormalities most commonly involve 14q32 (i.e., IGH) rearrangements, deletions at 1p, 13q, 11q22, and 17p, and duplications of the long arm of chromosome 1q. Chromosome changes are of diagnostic importance and provide critical prognostic information in multiple myeloma. ■ The t(14;20)(q32;q11.2) abnormality occurs recurrently only in multiple myeloma; thus, it serves as a diagnostic cytogenetic marker. ■ At least five recurrent IGH translocation partners have been identified in multiple myeloma. These are 11q13 (CCND1 in 15% to 20%), 4p16 (MMSET and FGFR3 in 10% to 20%), 16q23 (MAF in 5%), 6p21 (CCND3 in 3%), and 20q11.2 (MAFB in 2%). The following IGH translocations are associated with short progression-free survival and decreased overall survival: t(4;14), t(14;16), and t(14;20). ■ Hyperdiploidy in multiple myeloma predicts a good prognosis. Similarly, among the IGH translocation partners, the t(11;14)(q13;q32) translocation predicts a good prognosis. ■ The structural alterations involving the 17p13 (i.e., TP53) deletion and the 1q21 duplication predict an adverse outcome. These cytogenetic changes frequently occur with the adverse group of IGH translocations. ■ Thus, the specific cytogenetic aberrations can be used to define the prognostic classes in multiple myeloma.5,8 27. A. High-grade B-cell non-Hodgkin lymphoma (B-NHL). Major points of discussion ■ The t(8;14)(q24;q32) translocation and its variants, t(2;8)(p11.2;q24) and t(8;22)(q24;q11.2), are common in BL but can occur in almost any type of mature B-cell neoplasm. ■ The t(2;8)(p11.2;q24) translocation, involving rearrangement of the IGK gene with the MYC gene, are seen in approximately 5% of cases of BL and rarely in DLBCL. The t(2;8) translocation results in juxtaposition of the IGK enhancer to the MYC oncogene and deregulates the latter’s expression. ■ The t(2;8)(p11.2;q24) translocation in DLBCL is thought to be a secondary change associated with a complex karyotype. Most recurrent chromosome aberrations in complex karyotypes are gains of chromosomes X, 1q, 3, 7, 12, and 18; losses of chromosomes 6q, 13, 15, and 17; and are associated with aggressive behavior. ■ There is molecular heterogeneity regarding the breakpoints in the MYC gene between t(8;14) and t(8;22)/t(2;8). The MYC breakpoints in t(2;8) and t(8;22) are telomeric to MYC; in t(8;14) the breakpoint in MYC is centromeric.6,22,42 28. A. 46,XY,del(5)(q13q33). Major points of discussion ■ The incidence of MDS increases with age; most patients are over 60 years of age. Approximately 10% to 15% of MDS cases follow treatment (therapy-related MDS [t-MDS]) with chemotherapy and radiation for both neoplastic as well as benign disorders. ■ The WHO classification of MDS is based on bone marrow, histology, blast count, and cytogenetic findings. The cytogenetic evaluation of a bone marrow sample is valuable in assessing the prognosis, the risk for progression to acute leukemia, and survival in MDS patients. ■ A normal karyotype is found in 30% to 60% of patients with MDS depending on the method used (karyotype, FISH, aCGH, or SNP array) to detect chromosome abnormalities. The international MDS risk assessment workshop found that patients with a normal karyotype fell within the favorable risk group. The median survival for these patients is 3.8 years. The time to progression to acute leukemia of 25% of this cohort was 5.6 years. ■ A complex karyotype is found in approximately 20% of patients with primary MDS and in more than 90% of patients with t-MDS. A complex karyotype is defined as the presence of three or more chromosome abnormalities. The majority of these cases show abnormal chromosomes 5 or 7. A complex karyotype is associated with poor prognosis in MDS.25 29. A. Astrocytomas. Major points of discussion ■ A wide variety of cytogenetic abnormalities are found in tumors of the nervous system. ■ Astrocytomas are the best-studied adult nervous system tumors. Common gross genetic abnormalities include gain of chromosome 7 and loss of chromosome 10. Gain of epidermal growth factor receptor function is common in high-grade astrocytomas. ■ Medulloblastomas are primitive neuroectodermal tumors and are most commonly seen in children. The most common abnormality is isochromosome 17, which results in deletion of the short arm of chromosome 17p containing the REN and TP53 genes and gain of the long arm of chromosome 17q. ■ In meningiomas, loss of chromosome 22 and deletion 22q are common cytogenetic abnormalities. Patients with neurofibromatosis type II (NF2) have a predisposition to meningiomas. ■ Concomitant deletions of 1p and 19q are the characteristic cytogenetic abnormalities in oligodendrogliomas. Loss of both 1p and 19q correlates with a better response to chemotherapy and radiotherapy. ■ Ependymomas are typically benign. These tumors show deletions of chromosomes 19 and 22; otherwise, they have a relatively normal karyotype. 30. A. Chromophobe-type RCC. Major points of discussion ■ Major classes of RCC are clear cell, papillary, chromophobe, oncocytoma, collecting duct carcinomas, sarcomatoid transformation in RCC, and mucinous tubular and spindle cell carcinoma. Although most RCCs are sporadic, syndromic forms have been reported for each of the major histologic types. ■ Cytogenetic alterations are highly specific to each major histologic type of RCC and are of diagnostic relevance. ■ Clear cell RCC arising from mature renal tubular cells represents approximately 75% of all RCCs. Clear cell RCC is characterized by deletions on chromosome 3p. Several common regions of deletion at 3p14, 3p21, and 3p25 were identified. The 3q21 deletion is obligatory for classification as a clear cell RCC. Additional abnormalities include partial trisomy of 5q and gain of chromosomes 12 and 20. ■ Papillary RCC represents about 10% of all RCCs. Trisomy or tetrasomy of chromosome 7 and trisomy 17 are characteristic features of papillary RCC. Gain of chromosomes 12, 16, and 20 is present in aggressive tumors. ■ Chromophobe-type RCCs represent 5% of all RCCs. Cytogenetically, chromophobe RCC exhibits a modal chromosome number of 38-39 with frequent losses of chromosomes 1, 2, 6, 10, 13, 17, and 21. ■ Renal oncotyoma is a benign neoplasm. Chromosome abnormalities are seen in approximately 50% of oncocytomas. Loss of chromosomes 1 and 14, and loss of the Y chromosome in males, are seen in one subset of tumors. In a second subset, translocations at 11q13 are seen;. 11q13 rearranges with many different chromosomes in this setting. ■ Mucinous tubular and spindle cell carcinoma is a distinct form of RCC. Cytogenetic characteristics of very few cases have been reported. Loss of chromosomes 1, 14, and 15 has been described.12,28 31. A. Embryonal rhabdomyosarcoma. Major points of discussion ■ The t(11;22)(q24;q12) translocation results in rearrangement of the EWS gene at 11q24 and the FLI1 gene at 22q12. The translocation results in the formation of the ESW-FLI1 chimeric protein. ■ Approximately 5% of Ewing sarcomas result from variant translocations involving the EWS gene at 11q24, resulting in t(21;22)(q12;q12)[EWS-ERG] and t(7;22)(p22;q12) [EWS-ETV1].3,44 32. A. Chronic myeloid leukemia (CML), predicting a poor prognosis. Major points of discussion ■ The chromosome changes in B-cell ALL play an important role with regard to diagnostic classification and prognosis prediction. ■ Ploidy groups in ALL represent well-established cytogenetic entities comprising low hyperdiploidy (46 to 49 chromosomes), high hyperdiploidy (51 to 65 chromosomes), near-triploidy (66 to 79 chromosomes, near-tetraploidy (84 to 100 chromosomes), hypodiploidy (45 chromosomes), low hypodiploidy (31 to 39 chromosomes), and near-haploidy (25 to 29 chromosomes). ■ Each ploidy group predicts chemotherapy responses in ALL. Overall, the hyperdiploid ALL group responds well to standard chemotherapy compared with patients in the hypodiploid and near-haploid groups. ■ One third of children presenting with ALL harbor the high-hyperdiploidy karyotype (51 to 65 chromosomes). Children with high-hyperdiploid ALL respond well to standard chemotherapy regimens. Event-free survival rates are up to 80% at 5 years for this group of patients. ■ High-hyperdiploid ALL is characterized by a nonrandom gain of chromosomes X, 4, 6, 10, 14, 17, 18, and 21. However, involvement of other additional chromosomes can also be seen. ■ Within the high-hyperdiploid karyotype group, other variables such as gender, age, individual trisomies, and modal chromosome numbers can affect the outcome. For example, girls aged 1 to 9 years with trisomy 18, in particular, are reported to have the best event-free survival.24,29,46 33. A. The t(15;17)(q22;q21) translocation is commonly seen in MDS. Major points of discussion ■ The t(15;17) translocation is the sole cytogenetic abnormality in approximately 75% of cases. Trisomy 8 is the most common secondary change in 10% to 15% of cases. ■ Despite the remarkable specificity of t(15;17)(q22;q21) in APL, this translocation can rarely present as a secondary change during chronic myelocytic leukemia in blast crisis. ■ The t(15;17)(q22;q21) translocation results in the retinoic acid receptor α (RARA) gene on 17q22 fusing with a nuclear regulatory factor gene (i.e., the promyelocytic leukemia or PML gene) on 15q22, thereby giving rise to the PML/RARA fusion gene, which mediates the leukemogenic process. ■ APL with the t(15;17)(q22;q21) translocation exhibits a particular sensitivity to treatment with ATRA, which acts as differentiating agent. Response of APL treated with ATRA and other combinations of drugs (e.g., anthracyclines, arsenic trioxide) is highest compared with any other acute myelocytic leukemia subsets. ■ Variant translocations involving 17q22 (RARA) and NPM1 at 5q35, NUMA1 at 11q13, and ZBTB16 (previously known as PLZF) at 11q23 are seen in fewer than 2% of cases of APL.15,45 34. A. Chromophobe-type RCC. Major points of discussion ■ Major classes of RCC are clear cell, papillary, chromophobe, oncocytoma, collecting duct carcinomas, sarcomatoid transformation in RCC, and mucinous tubular and spindle cell carcinoma. Although most RCCs are sporadic, syndromic forms have been reported for each of the major histologic types. ■ Cytogenetic alterations are highly specific to each major histologic type of RCC and are of diagnostic relevance. ■ Clear cell RCC arising from mature renal tubular cells represents approximately 75% of all RCCs. Clear cell RCC is characterized by deletions on chromosome 3p. Several common regions of deletion at 3p14, 3p21, and 3p25 were identified. The 3q21 deletion is obligatory for classification as a clear cell RCC. Additional abnormalities include partial trisomy of 5q and gain of chromosomes 12 and 20. ■ Papillary RCC represents approximately 10% of all RCCs. Trisomy or tetrasomy of chromosome 7 and trisomy 17 are characteristic features of papillary RCC. Gain of chromosomes 12, 16, and 20 are present in aggressive tumors. ■ Chromophobe RCC represents approximately 5% of all RCCs. Cytogenetically, chromophobe RCC exhibits a modal chromosome number of 38-39 with frequent losses of chromosomes 1, 2, 6, 10, 13, 17, and 21. ■ Renal oncotyoma is a benign neoplasm. Chromosome abnormalities are seen in approximately 50% of oncocytomas. Loss of chromosomes 1 and 14, and loss of the Y chromosome in males, are seen in one subset of tumors. In a second subset, translocations at 11q13 are seen; 11q13 rearranges with many different chromosomes in this setting. ■ Mucinous tubular and spindle cell carcinoma is a distinct form of RCC. Cytogenetic characteristics of very few cases have been reported. Loss of chromosomes 1, 14, and 15 has been described. 35. A. Clear cell RCC. Major points of discussion ■ The 5-year survival and disease-free progression of RCCs varies by subtype: chromophobe RCC (100% and 94%), papillary RCC (86% and 88%), clear cell RCC (76% and 70%), and RCC unclassified (24% and 18%). Oncocytomas are benign neoplasms with very low risk of metastasis. ■ SNP arrays and aCGH could resolve morphologically challenging renal tumors. These genomic methods are becoming the method of choice for comprehensive evaluation of chromosomal imbalances in RCC. ■ Conventional cytogenetics is the method of choice when assessing for nonrecurrent balanced translocations, such as those present in familial non–von Hippel-Lindau clear cell RCC. FISH can be used to detect the presence of recurrent chromosomal translocations, such as those present in pediatric tumors involving the Xp11.2 locus. ■ Cytogenetic analysis does not provide distinction between papillary adenomas and low-grade RCCs because of overlapping combinations of numerical chromosome abnormalities. Tumor diameter remains an important diagnostic criterion.28,30 36. A. 45,XX,t(13;21)(p10;p10). Major points of discussion ■ The breakpoints primarily occur in the short arms, resulting in dicentric chromosomes. ■ Breaks may also occur in one short arm and one long arm of the participating chromosomes. This results in a monocentric rearrangement. ■ The formation of the Robertsonian translocation effectively creates a derivative chromosome, which consists primarily of the long arms of the two participating acrocentric chromosomes. ■ The formation of the Robertsonian translocation also produces a derivative chromosome, which consists of the short arms of the two participating acrocentric chromosomes. This small derivative chromosome is simultaneously lost, resulting in a net chromosome number of 45. Because the short arms of the acrocentric chromosomes contain a superfluous amount of ribosomal gene repeats, loss of this small derivative fragment is clinically insignificant as there are sufficient ribosomal genes on the remaining acrocentric short arms. ■ Either “rob” or “der” can be used to describe the Robertsonian translocation; that is21,54: • 45,XX,rob(13;21)(q10;q10) 37. A. Synovial sarcoma. Major points of discussion ■ The t(X;18) is a characteristic finding in synovial sarcomas. A third of these tumors show t(X;18) as a sole chromosome abnormality; the remaining show an unbalanced derivative chromosome X, resulting from t(X;18) and other complex chromosome abnormalities. Cytogenetic and molecular studies indicate that patients with simple karyotypes and the SS18-SSX2 fusion have a better clinical outcome than patients with a complex tumor karyotype and the SS18-SSX1 fusion. ■ Chromosome analysis in desmoplastic round cell tumors usually shows a near-diploid karyotype with a few numerical and structural aberrations. Identification of t(11;22) or the EWSR1-WT1 fusion transcript is useful in the differential diagnosis of small cell round cell tumors and may show higher sensitivity than immunohistochemical analyses. ■ Although no structural chromosome abnormalities are seen in embryonal rhabdomyosarcoma, gain of several chromosomes (i.e., chromosomes 2, 8, 12, 13, 1q, 7q, 11q) and loss of chromosomes 1p, 9p, 14q, 15q, and 17p are common. Patients with embryonal rhabdomyosarcomas have a better survival than those with alveolar rhabdomyosarcoma. ■ In alveolar rhabdomyosarcoma, t(2;13) and t(1;13) are often present in duplicate or triplicates; secondary numerical and structural abnormalities are common and the karyotypes are usually complex. Tumors with t(2;13), expressing PAX3-FOXO1A, have a higher propensity to metastasize to the bone marrow. 38. A. Embryonal rhabdomyosarcoma. Major points of discussion ■ Synovial sarcoma is characterized by the presence of a reciprocal balanced translocation between chromosome regions Xp11.2 and 18q11.2 [t(12;16)(q13;p11.2)] in more than 90% of the cases. ■ Rare variants have been described involving Xp11.2 alone or 18q11.2 alone, with either involved in translocations with other chromosomes. ■ The molecular consequence of the t(X;18)(p11.2;q11.2) translocation is the rearrangement between synovial sarcoma translocation on chromosome 18 (i.e., SS18, or alternatively, SYT) with one of three related synovial sarcomas, X breakpoint genes (i.e., SSX1, SSX2, or SSX4), resulting in different fusion genes (i.e., SS18-SSX1, SS18-SSX2, or SS18-SSX4). ■ Some synovial sarcoma cases exhibit complex karyotypes and amplification of the 12q13 region, which are associated with worse overall survival.14,26,53 39. A. BL. Major points of discussion ■ The TCRA/D region is frequently involved in translocations in T-PLL and in HTLV + adult T-cell lymphoma/leukemia. TCRA/D genes are also rarely involved in translocation rearrangements in T-cell ALL, ataxia telangiectasia, and leukemias of B lineage. ■ TCRA/D gene translocations are seen as inv(14)(q11.2q32) or in t(14;14)(q11.2;q32) translocations, partnering with the T-cell leukemia/lymphoma 1 (TCL1) gene at 14q32. ■ The genetic hallmark of T-PLL is inv(14)(q11.2q32), which is present in up to 80% of cases. Its variant, t(14;14)(q11.2;q32), is detected in approximately 10% of T-PLL cases. In both types of chromosome aberrations, the TCL1 gene at 14q32 is juxtaposed next to the TCRA/D locus at 14q11.2, resulting in up-regulation of TCL1 expression. ■ The TCR-associated translocations are regarded as the primary oncogenic events in T-PLL. ■ T-PLL is generally considered a very aggressive neoplasm among T-cell lymphomas, and they are poorly responsive to chemotherapy.18,50,62 40. A. 47,XX,+18. Major points of discussion ■ Trisomy 18 occurs with a frequency of 1:3333 to 1:6000 births. ■ Although trisomy 18 infants can survive to term, spontaneous abortion is the more likely outcome; thus, the prevalence of this chromosome abnormality is greater at the time of chorionic villus sampling and decreases all the way to term. ■ The median postnatal survival time is 14.5 days. ■ Only 5% to 10% of trisomy 18 newborns survive the first year. ■ Those who survive have severe mental deficiency.21,33,43,63 41. A. A 30-year-old woman with a 46,XX karyotype. Major points of discussion ■ Robertsonian translocation carriers are increased risk for trisomy or monosomy because of unbalanced segregation at meiosis. ■ Advanced maternal age is the most important etiologic factor in risk for aneuploidy and human reproductive failure. Association of advanced maternal age and Down syndrome was known even before the chromosomal basis of Down syndrome was discovered. ■ Women with the karyotype 45,XX,t(21;21)(q10;q10) can only have only trisomy 21 or monosomy 21 conceptions. Conceptions with monosomy 21 result in spontaneous abortions. A trisomy 21 conception may survive and result in a live birth with the infant having Down syndrome. ■ Female carriers of the Robertsonian translocation (14;21) are at increased risk, a 10% to 15% risk of trisomy 21. The risk for male carriers is very different; it is less than 1%.49,52,58
Cytogenetics
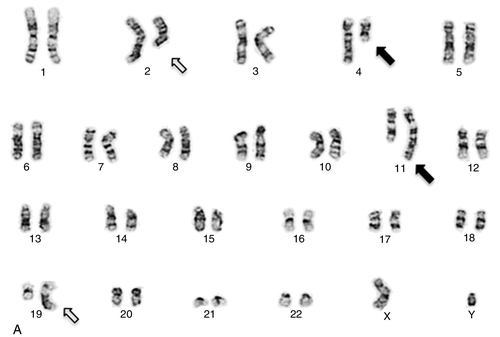
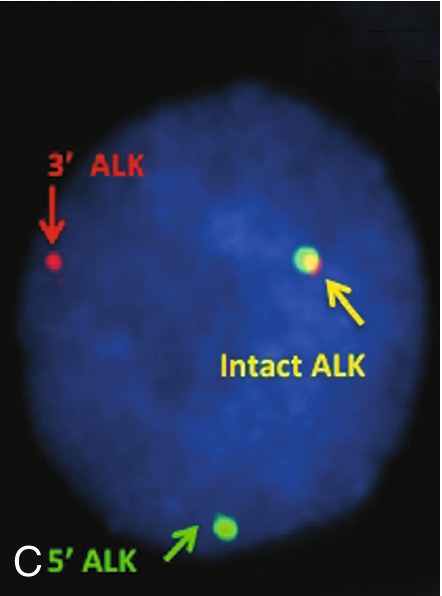
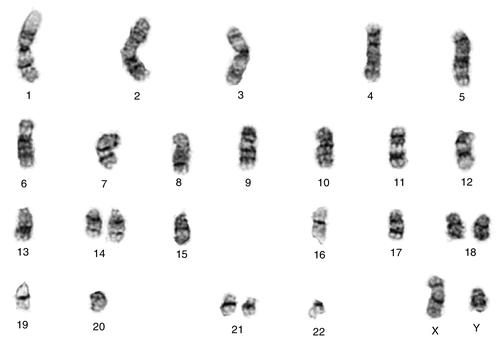
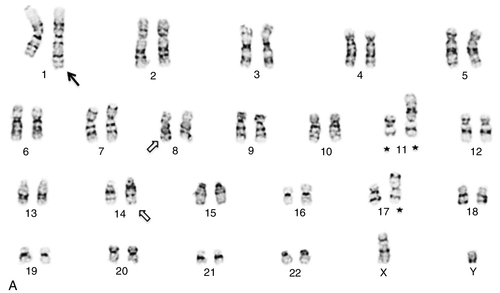
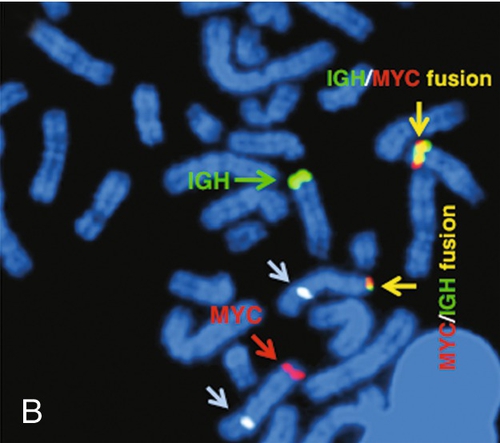
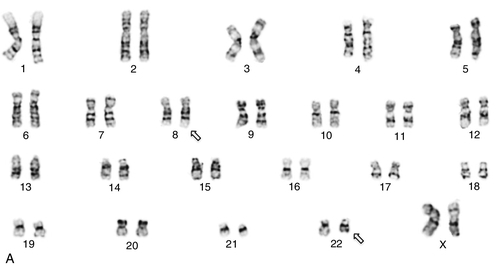
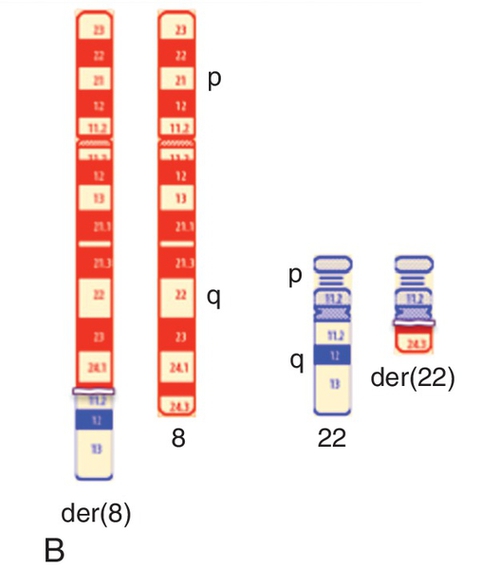
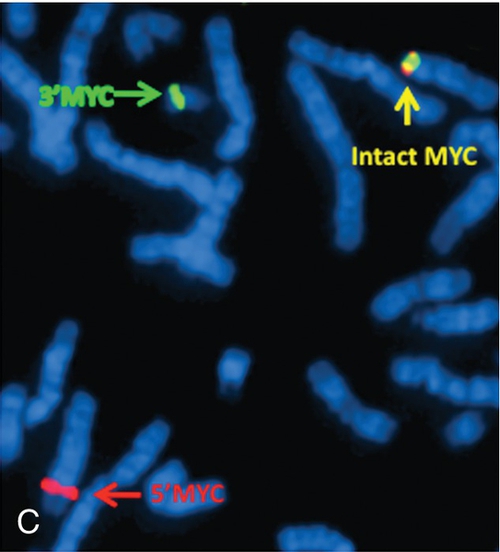
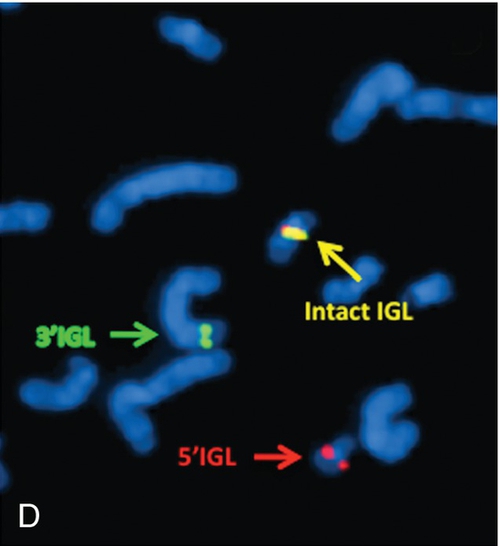
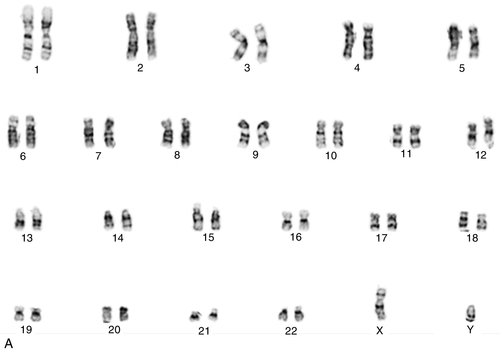
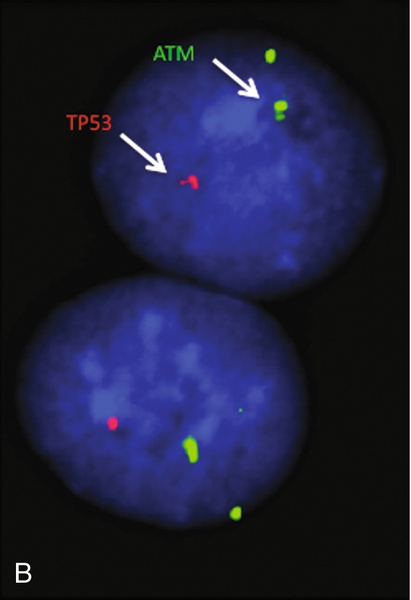
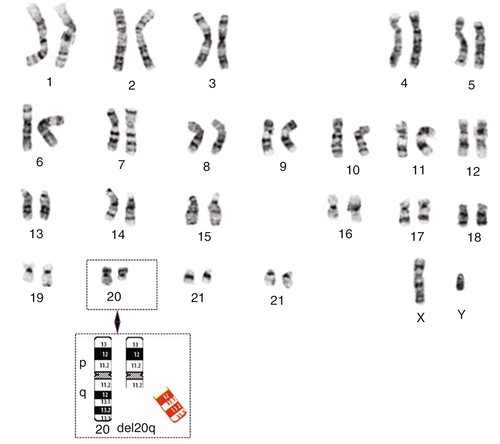
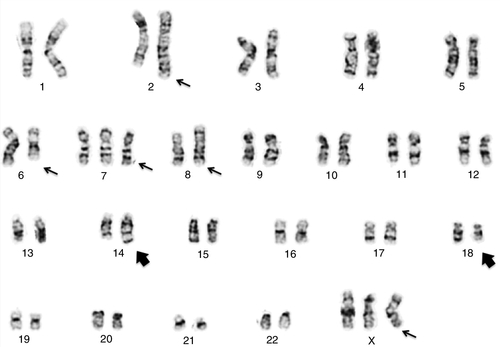
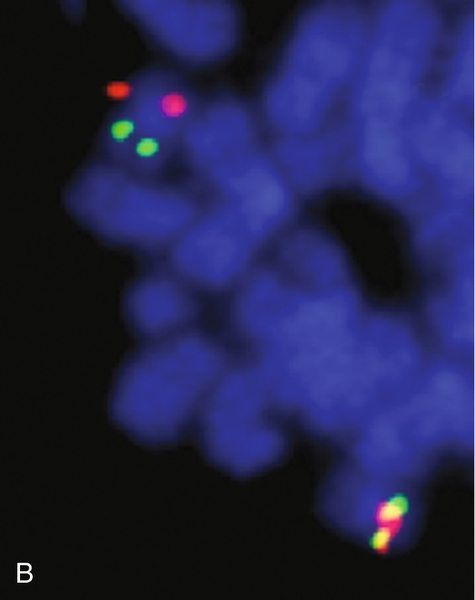
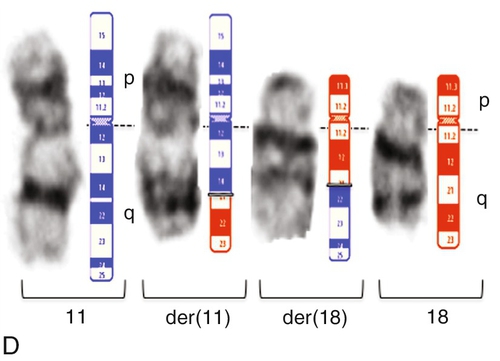
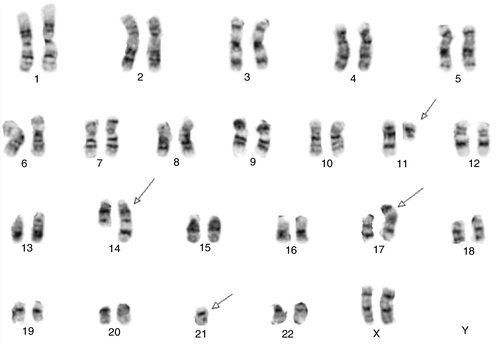
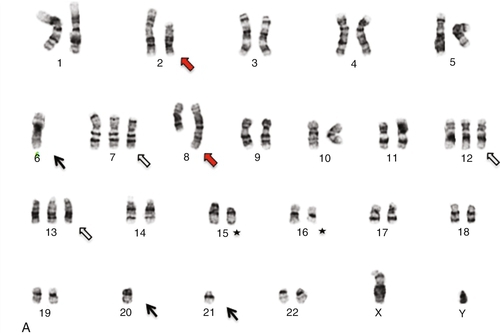
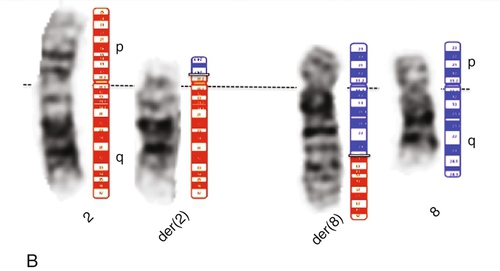
45, X,–Y,trp(1)(q22q31),add(12)(q13),ins(13:?)(q14;?),t(14;20)(q32;q11.2),add(14)(q32)[10]/46,XY[10].
Based on the cytogenetic breakpoint at 14q32, FISH analysis was performed using a IGH dual-color break-apart probe and IGH was found to be rearranged, as shown in Figure 23-15, B. Based on the results of these analyses, what is the best interpretation?
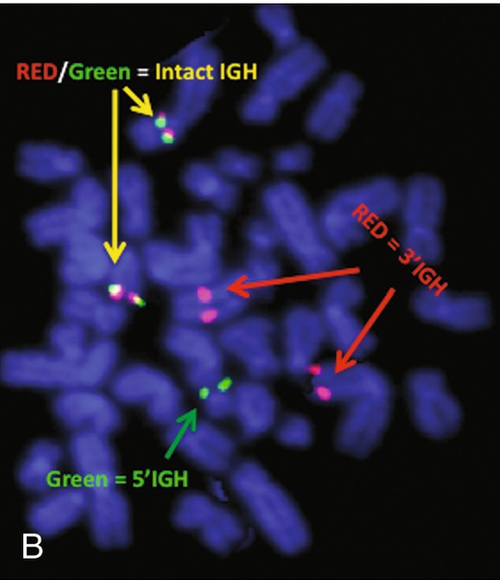
Rationale: t(8;14)(q24;q32), t(8;22)(q24;q11), and t(2;8)(p11;q24) are the characteristic chromosome translocations in BL. The t(4;11) translocation is not seen in BL.
B. Myelodysplastic syndrome (MDS) with a good prognosis.
Rationale: The chromosome changes frequently seen in MDS are del(5q)/monosomy 5, del(7q)/monosomy 7, del(20q)/monosomy 20, and trisomy 8. The t(4;11) translocation is not seen in MDS.
C. Acute lymphoblastic leukemia (ALL) with a poor prognosis.
Rationale: The t(4;11)(q21;q23) translocation and the associated MLL rearrangement are seen in ALL and the prognosis is very poor.
D. ALL with a good prognosis.
Rationale: Although the t(4;11)(q21;q23) translocation and the associated MLL rearrangement are seen in acute lymphoblastic leukemia, this translocation is not associated with a good prognosis.
E. Acute promyelocytic leukemia (APL) with a good prognosis.
Rationale: APL is associated with chromosome translocation t(15;17)(q22;q21). The t(4;11) translocation is not seen in APL.
Rationale: No specific chromosome translocation has been identified in NK-cell neoplasms. However, deletions of 6q and 13q are frequently seen.
B. Anaplastic large cell lymphoma (ALCL).
Rationale: The immunophenotype in this case is consistent with an ALCL. A balanced translocation between chromosome regions 2p23 and 5q35 [t(2;5)(p23;q35)], resulting in an ALK rearrangement, is a classic feature of ALCL.
C. Hepatosplenic T-cell lymphoma.
Rationale: An isochromosome of the long arm of chromosome 7 [i(7)(q10)] is seen in most cases of hepatosplenic T-cell lymphoma.
D. T-cell prolymphocytic leukemia (T-PLL).
Rationale: The inv(14)(q11.2q32) abnormality is seen in most cases (~ 65%) of T-PLL.
E. Angioimmunoblastic T-cell lymphoma.
Rationale: The classic cytogenetic feature of angioimmunoblastic T-cell lymphoma is the presence of unrelated clones. Trisomies of chromosomes 3, 5, and X are seen in most cases.
Rationale: Specific balanced chromosome translocations, such as t(8;14)(q24;q32), t(8;22)(q24;q11), and t(2;8)(p11;q24,), are the characteristic of BL. A near-haploid karyotype is not seen in BL.
B. MDS, predicting a good prognosis.
Rationale: The chromosome changes frequently seen in MDS are del(5q)/monosomy 5, del(7q)/monosomy 7, del(20q)/monosomy 20, and trisomy 8. A near-haploid karyotype is not seen in MDS.
C. ALL, predicting a poor prognosis.
D. ALL, predicting a good prognosis.
Rationale: A near-haploid karyotype is a characteristic feature of a subset of patients with ALL and is associated with a short complete remission and a poor prognosis.
E. APL, predicting a good prognosis.
Rationale: APL is associated with the following specific chromosome translocation: t(15;17)(q22;q21). A near-haploid karyotype is not seen in APL.
Rationale: Morphologically, the majority of t(8;21)-positive AML patients display Auer rods and dysplastic features of the granulocytes. This translocation results in RUNX1-RUNX1T1 fusion and is associated with a favorable prognosis.
B. 46,XY,inv(16)(p13q22).
Rationale: AML cases with inv(16) are morphologically classified with a variable number of eosinophils, often with nuclear blebs and Auer rods. This inversion results in a CBFB/MYH11 fusion and is considered a favorable prognosis marker in AML.
C. 46,XY,t(4;11)(q21;q23).
Rationale: This translocation is strongly associated with ALL. However, this translocation has been described in few cases of AML. AML with t(4;11) is more common in the pediatric population, often in infant leukemias. Characteristic features of AML patients with this translocation include monocytic morphology, leukocytosis, previous chemotherapy, and poor prognosis.
D. 46,XY,t(15;17)(q24;q21).
Rationale: APL is characterized by a preponderance of abnormal promyelocytes with conspicuous granules and bundles of Auer rods. The t(15;17) translocation is considered pathognomonic for APL.
E. 46,XY,inv(3)(q21q26).
Rationale: Inversion of chromosome 3 is associated with dysplastic megakaryocytes and increased platelets. This inversion results in the activation and overexpression of the EVI1 gene, which has been described as an independent prognostic factor within the intermediate-risk karyotypic group.
Rationale: The t(8;14)(q24;q32) translocation, resulting in MYC rearrangement with the IGH locus, is the characteristic chromosomal translocation in BL. The 1q duplication, as seen in this case, is commonly seen in progressed BL.
B. Hodgkin disease.
Rationale: In contrast to non-HLs, specific chromosome translocations have not been identified in Hodgkin disease.
C. Multiple myeloma.
Rationale: The 14q32 region involving the IGH gene is frequently partnered with 11q13 (the cyclin D1 gene), 4p16 (the FGFR3 gene), 6p25 (the cyclin D3 gene), 16q23 (the MAF gene), and 20q11 (the MAFB gene), thereby forming balanced reciprocal translocations. Other changes frequently seen in multiple myeloma are 11q23 and 6q deletions, 1q duplications, and trisomies of chromosomes 3, 5, 7, 9, 11, 15, 19, and 21.
D. Mucosa-associated lymphoid tissue (MALT) lymphoma.
Rationale: The t(11;18)(q21;q21) balanced chromosome translocation is primarily detected in MALT lymphomas.
E. MZL.
Rationale: The three distinct clinicopathologic subtypes of MZL exhibit no characteristic chromosome changes, although trisomies 3 and 18, 7q deletions, and 14q32 rearrangements with various partners are frequently seen.
Rationale: The t(8;22)(q24;q11.2) translocation, resulting in MYC rearrangement with the IGL locus, is the characteristic chromosome translocation seen in approximately 10% of BL cases.
B. MZL.
Rationale: The three distinct clinicopathologic subtypes of MZL exhibit no particular diagnostic chromosome changes. However, the following can be seen: trisomies 3 and 18, 7q deletions, and 14q32 rearrangements with various partners.
C. Multiple myeloma.
Rationale: The 14q32 region involving the IGH gene is frequently partnered with 11q13 (the cyclin D1 gene), 4p16 (the FGFR3 gene), 6p25 (the cyclin D3 gene), 16q23 (the MAF gene), and 20q11 (the MAFB gene), thereby forming balanced reciprocal translocations. Other changes frequently seen in multiple myeloma are 11q23 and 6q deletions, 1q duplications, and trisomies of chromosomes 3, 5, 7, 9, 11, 15, 19, and 21.
D. MALT lymphoma.
Rationale: The t(11;18)(q21;q21) balanced chromosomal translocation is primarily detected in cases of MALT lymphoma.
E. AML.
Rationale: AML is not associated with t(8;22) translocations. The most frequently identified abnormalities in AML are t(15;17)(q22;q21), t(8;21)(q22;q22), 11q23 rearrangement, and inv(16)(p13q22).
Rationale: Standard G-band karyotype with no identifiable chromosome changes is not a useful predictor of prognosis.
B. The two copies of the ATM gene by interphase FISH (Figure 23-6, B) predict a good prognosis.
Rationale: Disomy of the ATM gene is a normal condition and is not indicative of any particular prognosis in CLL.
C. The G-band karyotype (Figure 23-6, A) and FISH (Figure 23-6, B) together do not predict any particular prognosis.
Rationale: Although a normal karyotype is not predictive of prognosis, deletion of one copy of TP53 predicts a poor prognosis in CLL.
D. Heterozygous deletion of the TP53 gene (Figure 23-6, B) predicts a poor prognosis.
Rationale: Deletion 17p13 (TP53) as a sole abnormality, or in combination with other chromosome aberrations, is associated with an unfavorable clinical outcome.
E. Heterozygous deletion of the TP53 gene (Figure 26-3, B) predicts a good prognosis.
Rationale: Deletion 17p13 (TP53) alone, or in combination with deletion of 11q22-23 (ATM), is associated with an unfavorable clinical outcome in CLL.
B. 15% to 30%.
Rationale: These incidences are far too low.
C. 30% to 45%.
Rationale: This incidence is closer to the figures quoted in earlier studies performed in the 1980s that indicated an incidence of 50%.
D. 60% to 70%.
Rationale: Although studies in the 1970s indicated the incidence of chromosome abnormalities in spontaneous abortions to be approximately 50%, more recent studies demonstrated that by selectively using chorionic villi, or by meticulously dissecting out fetal parts, the percentage of cases with abnormal karyotypes increases to 60% to 70%. This is primarily due to a reduction of maternal cell contamination (by meticulous dissection of fetal tissues) as well as improvements in culture success rates.
E. 75% to 80%.
Rationale: This incidence is far too high.
Rationale: Although the alternating light and dark bands observed on each chromosome appear like a “bar code,” the correct term to use is “banding pattern,” not “bar code.” The centromere size is not distinguishable from one chromosome to the next, and only a few chromosomes have discrete heterochromatic blocks that vary in size in the population.
B. The size of the chromosome, the banding pattern, and the length of the telomere.
Rationale: The actual telomere is a submicroscopic, highly repetitive DNA sequence and, therefore, cannot be observed by light microscopy. The size of the chromosome and the banding pattern are used to uniquely identify each chromosome.
C. The length of the telomere, the location of the centromere, and their specific bar code.
Rationale: Although the alternating light and dark bands observed on each chromosome appear like a bar code, the correct term to use is banding pattern, not bar code. The position of the centromere is a distinguishing feature of a chromosome and is one of the factors used for identifying chromosomes.
D. The position of the centromere, the size of the chromosome, and the banding pattern.
Rationale: The position of the centromere, the size of the chromosome, and the banding pattern are the primary factors used to identify chromosomes. The centromere position of each chromosome is not variable and the size of each chromosome remains constant in relation to all the others. Each chromosome also has a unique banding pattern.
E. The location of the heterochromatin, their specific bar code pattern, and the location of the centromere.
Rationale: Only a few chromosomes have discrete heterochromatic blocks and in all cases, these heterochromatic blocks are located just below the centromere. Although the alternating light and dark bands observed on each chromosome appear like a bar code, the correct term to use is banding pattern, not bar code. The position of the centromere is a distinguishing feature of a chromosome and is one of the factors used for identifying chromosomes.
Rationale: Isoleucine is a stable amino acid that can be added to the culture medium when it is being formulated.
B. l-Lysine.
Rationale: Lysine is a stable amino acid that can be added to the culture medium when it is being formulated.
C. l-Valine.
Rationale: Valine is a stable amino acid that can be added to the culture medium when it is being formulated.
D. l-Leucine.
Rationale: Leucine is a stable amino acid that can be added to the culture medium when it is being formulated.
E. l-Glutamine.
Rationale: l-Glutamine is unstable and breaks down on storage to d-glutamine, a form that cannot be used by cells. l-Glutamine may be kept frozen to retain its stability and should only be added to culture medium just prior to use.
Rationale: Sample mix-ups can be seen in clinical laboratories; however, the mixing of samples is not common.
B. The 69,XXY cells are an artifact of tissue culture and the POC contains only normal female cells.
Rationale: Cells with a 69,XXY karyotype are not artifacts of tissue culture. Triploidy is found in approximately 2% of spontaneous abortions.
C. The POC has a 69,XXY karyotype and the cells with the 46,XX karyotype are maternal in origin.
Rationale: Overgrowth of maternal cells in culture is common from abortion/miscarriage specimens. Chromosome analysis from these specimens requires diligent cleaning away of adherent maternal tissue.
D. This is a twin pregnancy: One twin has a 69,XXY karyotype and the other twin has a 46,XX karyotype.
Rationale: This is a possibility; however, overgrowth of maternal cells in culture is the most likely scenario.
E. This represents true triploidy mosaicism; the fetus has two cell lines (69,XXY and 46,XX).
Rationale: This is a possibility; however, true triploid mosaicism is very rare and very few cases are described in the literature.
Rationale: Deletion 20q as an isolated abnormality is a common recurring change in a spectrum of myeloid malignancies, such as MDS and AML.
B. Deletion 20q as the sole abnormality is associated with MDS and predicts a good prognosis.
Rationale: Deletion 20q is seen in 5% to 7% of cases of both isolated MDS and treatment-related MDS. The International Prognosis Scoring System (IPSS) classified deletion 20q as a favorable prognostic marker when it occurs as an isolated abnormality in cases of MDS.
C. Deletion 20q as the sole abnormality is diagnostic of a specific subtype of AML and predicts a poor prognosis.
Rationale: Deletion 20q is not specific to any particular FAB subtype of AML and, as an isolated abnormality, is seen in less than 1% of AML patients. In this setting, it is not a favorable cytogenetic change, as it is in MDS.
D. Deletion 20q as the sole abnormality predicts a poor prognosis in MDS.
Rationale: According to the IPSS, deletion 20q as an isolated abnormality predicts good prognosis.
E. Deletion 20q in a complex karyotype predicts a good prognosis in MDS.
Rationale: Deletion 20q in the setting of a complex karyotype is of less favorable prognostic value.
Rationale: The absence of Down syndrome in multiple prior pregnancies diminishes the likelihood of inherited forms of Down syndrome due to familial balanced rearrangements. It does not diminish the risk for the most common mechanism (nondisjunction leading to simple trisomy of chromosome 21). The risk for nondisjunction increases with maternal age and the risk for this couple would actually be higher than for any of their previous pregnancies, simply because the mother is older.
B. High, as most Down syndrome cases are inherited.
Rationale: Most cases of Down syndrome (~ 95%) are not inherited and are due to nondisjunction leading to simple trisomy of chromosome 21.
C. The same as the age-related risk of the mother.
Rationale: The absence of Down syndrome in multiple prior pregnancies diminishes the likelihood of inherited forms of Down syndrome due to familial balanced rearrangements. It does not diminish the risk for the most common mechanism (nondisjunction leading to simple trisomy of chromosome 21). The risk for nondisjunction increases with maternal age and the risk for this couple would be equal to the mother’s age-related risk.
D. High, because the mother of the child with Down syndrome was 22 when the child was born.
Rationale: Although the risk of having a child with Down syndrome increases with maternal age, Down syndrome children are still born to younger mothers.
E. Low, because the cousin is on the father’s side of the family.
Rationale: The risk of a familial form of Down syndrome (i.e., translocation) is not influenced by which side of the family the index case occurred.
Rationale: This answer corresponds to the karytoype of a normal male fetus. The signal pattern with this karyotype would be:
Rationale: This karytoype indicates a male fetus with a Robertsonian translocation. The signal pattern with this karyotype would be:
Rationale: This karyotype indicates a male fetus with a Robertsonian translocation and trisomy 21. The signal pattern with this karyotype would be:
Rationale: The observed signal pattern is consistent with this karyotype.
E. 46,XY,der(13;21)(q10;q10),+18.
Rationale: This karyotype indicates a male fetus with a Robertsonian translocation and trisomy 18. The signal pattern with this karyotype would be:
Rationale: t(8;14)(q24;q32), t(8;22)(q24;q11), and t(2;8)(p11;q24) are the characteristic chromosome translocations in BL.
B. HL.
Rationale: In contrast to non-HLs, specific chromosome translocations have not been identified in HL.
C. Multiple myeloma.
Rationale: Both structural and numerical chromosome changes are seen in multiple myeloma. The 14q32 region involving the IGH gene is frequently partnered with 11q13 (the cyclin D1 gene), 4p16 (the FGFR3 gene), 6p25 (the cyclin D3 gene), 16q23 (the MAF gene), and 20q11 (the MAFB gene), thereby forming balanced reciprocal translocations. Other changes frequently seen in multiple myeloma are 11q23 deletions, 6q deletions, 1q duplications, and trisomies of chromosomes 3, 5, 7, 9, 11, 15, 19, and 21.
D. MALT lymphoma.
Rationale: The t(11;18)(q21;q21) balanced chromosome translocation is primarily detected in MALT lymphoma.
E. Follicular lymphoma (FL).
Rationale: t(14;18)(q32;q21) is a characteristic feature of FL and is used as a diagnostic marker for this type of lymphoma. Other frequently seen chromosome changes in high-grade FL are deletion of 6q and trisomies of chromosomes 7, 12, and X.
Rationale: The 46,XX karyotype reflects a normal female karyotype. However, the hybridization pattern shows one green and one red signal corresponding to a male chromosome composition.
B. 47,XXY.
Rationale: The 47,XYY karyotype is for a male who has two X chromosomes and a single Y chromosome, which is consistent with a clinical diagnosis of Klinefelter syndrome. The hybridization pattern shows one green and one red signal corresponding to one copy of the X chromosome and one copy of the Y chromosome.
C. 47,XX,+18.
Rationale: The 47,XX,+18 karyotype indicates a female with an extra copy of chromosome 18; that is, trisomy 18. The hybridization pattern shows one green, one red, and two aqua signals corresponding to one copy of the X chromosome, one copy of the Y chromosome, and two copies of chromosome 18.
D. 47,XY,+18.
Rationale: The 47,XY,+18 karyotype indicates a male with an extra copy of chromosome 18; that is, trisomy 18. The hybridization pattern shows one green, one red, and two aqua signals corresponding to one copy of the X chromosome, one copy of the Y chromosome, and two copies of chromosome 18.
E. 46,XY.
Rationale: The 46,XY karyotype reflects a normal male karyotype. If all the remaining chromosomes are normal, the expected hybridization pattern for the probes indicated above should be one green, one red, and two aqua signals corresponding to one copy of the X chromosome, one copy of the Y chromosome, and two copies of chromosome 18.
Rationale: “nuc ish” is always used for describing FISH results on interphase nuclei.
B. 46,XX.ish(CEP-X,CEP-Y)x1,(CEP-18) × 2.
Rationale: 46,XX.ish indicates that FISH has been performed on a metaphase spread. Probe results from the same hybridization are described in the same set of parentheses and not separately. The hybridization pattern in Figure 23-10 is also consistent with a male, not a female.
C. 46. nuc ish(CEP X,Y,18,18).
Rationale: Interphase FISH results do not indicate predicted chromosome number (unless traditional karyotyping has simultaneously been performed). Interphase FISH results should indicate the number of signals for each probe.
D. nuc ish(DXZ1x1,DYZ3x1,D18Z1 × 2).
Rationale: Interphase FISH results are designated by “nuc ish” followed by the number of signals for each probe. Probe results from the same hybridization are described in the same set of parentheses.
E. nuc ish(X,Y,18,18).
Rationale: Interphase FISH results should indicate the number of signals for each probe.
Rationale: Three signals are observed for all probes used in this assay. Given the ultrasound anomalies, the diagnosis of triploidy is the most likely explanation.
B. A fetus with tetraploidy.
Rationale: If the fetus has tetraploidy, four signals would be observed for each probe used in this assay.
C. A fetus with trisomy 13, trisomy 16, trisomy 18, trisomy 21, and trisomy 22.
Rationale: Although the fetus does indeed have trisomy of these chromosomes, multiple aneuploidy involving five chromosomes has not been reported prenatally. The three signals observed for each of these chromosomes are an indication that there is an extra set of all chromosomes; that is, triploidy.
D. A fetus with malignant transformation.
Rationale: Trisomy of chromosomes 13, 16, 18, 21, and 22 has not been described as a malignant event in chorionic villi obtained from a 10-week-old fetus.
E. Uncertain, because the multiple signals observed are more likely an indication of assay failure. A repeat FISH analysis is indicated.
Rationale: Multiple signals are commonly observed in FISH analyses and are not an indication of assay failure.
Rationale: Inversion (16)(p13.1q22), t(16;16)(p13.1;q22), and deletion 16q22 are variants of each other. These changes are pathognomonic of acute myelomonocytic leukemia.
B. Inversion 16 and CBFB gene rearrangement are diagnostic of a specific subtype of AML and predict a good prognosis.
Rationale: Inversion 16 and the associated CBFB rearrangement are diagnostic of AML with monocytic and granulocytic differentiation and abnormal eosinophils. This change predicts a better prognosis and results in a longer complete remission when treated with high-dose cytarabine.
C. Inversion 16 and CBFB gene rearrangement are diagnostic of a specific subtype of AML and predict a poor prognosis.
Rationale: Inversion 16 is diagnostic of AML M4Eo and predictive of a high complete remission rate; therefore, this predicts a good prognosis.
D. Inversion 16 and CBFB gene rearrangement are diagnostic of a specific subtype of AML but have no prognostic value.
Rationale: AML with inversion 16, and its other variants, is highly responsive to treatment; therefore, this is a prognostic indicator.
E. Inversion 16 and CBFB gene rearrangement are diagnostic of ALL.
Rationale: Inversion 16 is not seen in ALL. B-cell ALL exhibits other characteristic chromosome changes, which are of diagnostic and prognostic value.
Rationale: The t(8;14)(q24;q32) translocation, resulting in MYC rearrangement with the IGH locus, is the characteristic chromosome translocation in BL.
B. HL.
Rationale: In contrast to non-HLs, specific chromosome translocations have not been identified in HL.
C. FL.
Rationale: The t(14;18)(q32;q21) translocation is a characteristic feature of FL and is used as a diagnostic marker for this type of lymphoma.
D. MALT lymphoma.
Rationale: The t(11;18)(q21;q21) balanced chromosomal translocation is diagnostic of MALT lymphoma.
E. Splenic marginal zone lymphoma (SMZL).
Rationale: Although SMZL exhibits no characteristic chromosome changes, trisomies 3 and 18, 7q deletions, and 14q32 rearrangements are frequently seen.
Rationale: t(8;14)(q24;q32), t(8;22)(q24;q11), and t(2;8)(p11;q24) are the characteristic chromosome translocations in BL.
B. Mantle cell lymphoma.
Rationale: Mantle cell lymphoma presents with a t(11;14)(q13;q32) translocation in virtually all cases; this translocation is used as a diagnostic marker. Although t(11;14) is seen in rare cases of multiple myeloma and chronic lymphocytic leukemia, it is not a diagnostic feature of these lymphomas.
C. MZL.
Rationale: Chromosome alterations involving the gain of the long arms of chromosomes 3 and 18 are commonly seen in MZLs.
D. MALT lymphoma.
Rationale: The chromosome change t(11;18)(q21;q21) is primarily detected in MALT lymphomas.
E. Diffuse large B-cell lymphoma.
Rationale: Approximately 30% to 40% of diffuse large B-cell lymphoma (DLCBL) cases show abnormalities at the 3q27 region involving the BCL6 gene, with the IHC locus at 14q32 as the major translocation partner. Translocation t(14;18)(q32;q21) and deletion 6q are the other major chromosome changes in diffuse large B-cell lymphoma.
Rationale: DiGeorge syndrome is caused by a 1.5- to 3.0-Mb hemizygous deletion of the proximal long arm region of chromosome at 22q11.2. Haploinsufficiency of the TBX1 gene is believed to be responsible for most of the physical malformations. The principal clinical features include hypocalcemia arising from parathyroid hypoplasia, thymic hypoplasia, and outflow tract defects of the heart.
B. Cat-eye syndrome.
Rationale: Cat-eye syndrome is usually associated with an SMC derived from chromosome 22. The SMC is often dicentic and bisatellited and represents an inversion duplication of the genomic material from the proximal short arm to the proximal long arm of chromosome 22. There is great variability in the clinical features, especially the congenital malformations. The principal clinical features include coloboma of the iris, anal atresia with fistula, downslanting palpebral fissures, preauricular tags and/or pits, and the frequent occurrence of heart and renal malformations. Mental development may be near-normal or normal, with as many as 47% of cases falling within the normal range (IQ greater than 85).
C. Angelman syndrome.
Rationale: Angelman syndrome is a neurodevelopmental disorder characterized by mental retardation, movement or balance disorder, typical abnormal behaviors, and severe limitations in speech and language. Most cases are caused by the lack of a maternal contribution to the imprinted region on chromosome 15q11-q13. Approximately 70% of Angelman syndrome cases are due to a deletion of the critical genomic region on the maternally inherited chromosome at 15q11-13. Other mechanisms include mutation in the E3 ubiquitin-protein ligase gene (UBE3A) (5% to 10%), paternal uniparental disomy (2% to 5%), and an imprinting defect (2% to 5%). In the remainder of cases, the mechanisms are unknown.
D. Charcot-Marie-Tooth syndrome type 1A.
Rationale: Charcot-Marie-Tooth disease is a sensorineural peripheral polyneuropathy. It is the most common inherited disorder of the peripheral nervous system affecting approximately 1:2,500 individuals. Charcot-Marie-Tooth disease type 1A is caused by duplication of, or mutation in, the PMP22 gene located on the short arm of chromosome 17 at 17p12.
E. Beckwith-Wiedemann syndrome.
Rationale: Beckwith-Wiedemann syndrome is a pediatric overgrowth disorder involving a predisposition to tumor development. The classic features include exomphalos, macroglossia, and gigantism. Clinical presentation is highly variable. Beckwith-Wiedemann syndrome is caused by mutation or deletion of imprinted genes within the chromosome 11p15.5 region. Specific genes involved include CDKN1C, H19, and LIT1. Cytogenetically detectable abnormalities (duplication, inversion, or translocation) involving chromosome 11p15 are found in 1% or less of affected patients. Loss or gain of methylation at the imprinting center 2 (IC2) on the maternal chromosome account for roughly 50% and 5% of cases, respectively. Paternal uniparental disomy of 11p15.5 accounts for approximately 20% of cases.
Rationale: The presence of mosaicism has no bearing on the ability to identify the origin of a marker chromosome.
B. Commercial chromosome 22 probes are not approved by the Food and Drug Administration (FDA).
Rationale: Most commercial probes are not FDA approved, and this has no bearing on the ability to identify the origin of a marker chromosome.
C. Chromosomes 14 and 22 share common α-satellite sequences.
Rationale: Because more than 99% of markers are centric fragments of variable size, centromere probes are the best-suited probes to identify the origin of a marker chromosome. Every human centromere is composed of α-satellite sequences, and the commercial centromere probes are typically composed of these α-satellites sequences. The centromeres of most human chromosomes contain unique α-satellite sequences that can distinguish one chromosome from another. However, chromosomes 14 and 22 share common α-satellite sequences and, therefore, cannot be uniquely identified when using commercial centromeric probes.
D. There are no commercial probes for chromosome 22.
Rationale: There are multiple commercially available FISH probes that map to chromosome 22.
E. Most markers derived from chromosome 22 also contain chromosomal sequences from chromosome 18.
Rationale: See Major Points of Discussion.
Rationale: Marker chromosomes, derived from chromosome 15, that do not include the Prader-Willi/Angelman critical region (as shown by FISH in this case, which was negative for the SNRPN probe on the marker chromosome) are typically not associated with an adverse clinical phenotype. The finding of biparental inheritance of the normal chromosome 15 homologues effectively rules out a genomic disorder caused by uniparental disomy.
B. High, because chromosome 15 contains imprinted genes.
Rationale: Although chromosome 15 does contain imprinted genes, biparental inheritance of the normal chromosome 15 homologues effectively rules out a genomic disorder caused by uniparental disomy.
C. Negligible, because there are no imprinted genes on chromosome 15.
Rationale: Chromosome 15 does contain imprinted genes.
D. High, because the marker chromosome likely resulted from a rescue of a trisomy 15 fetus.
Rationale: Although the marker chromosome may have resulted from the rescue of a trisomy 15 fetus, the follow-up molecular studies revealed biparental inheritance of the normal chromosome 15 homologues, which effectively rules out the risk of a genomic disorder caused by uniparental disomy.
E. Unclear, because further molecular testing is required before accurate risk assessment can be made.
Rationale: In this clinical scenario, all the relevant testing has been performed to allow for an appropriate clinical interpretation.
Rationale: Balanced translocations are usually not associated with any clinical phenotype unless the breakpoints occur in a critical region of a gene, thereby disrupting gene function, or there is a concomitant microdeletion or microduplication at the breakpoint sites.
B. An unbalanced translocation that always involves the proximal long arm of chromosome 22.
Rationale: Unbalanced translocations are associated with a clinical phenotype. However, chromosome 15 needs to be involved, with a net loss of the proximal long arm region, in order to result in PWS.
C. An insertion of extra chromosomal material into the distal long arm region of chromosome 17.
Rationale: Extra material inserted into another chromosome is likely to be associated with a clinical phenotype. However, chromosome 15 needs to be involved, with net loss of the proximal long arm region, in order to result in PWS.
D. A duplication of chromosomal material at the proximal region of the long arm of chromosome 7.
Rationale: A duplication of the proximal 7q region is associated with the 7q11.23 duplication syndrome, which is characterized by facial dysmorphism and prominent speech delay.
E. A deletion on the proximal region of the long arm of chromosome 15.
Rationale: A deletion, specifically of the paternally inherited chromosome 15 at 15q11.2, is associated with a clinical diagnosis of PWS.
Rationale: Paint probes cannot detect microdeletion syndromes. Paint probes are used to detect chromosome translocations and to determine the origin of a marker chromosome.
B. Spectral karyotyping (SKY) using multicolor chromosome paint probes for all chromosomes.
Rationale: This method is mainly used to identify structural chromosome abnormalities such as translocations and marker chromosomes. Small deletions cannot be detected by SKY.
C. A locus-specific FISH probe for the commonly deleted region on chromosome 17.
Rationale: FISH using a locus-specific probe for the SMS critical region containing the RAI1 gene is used to detect this microdeletion on chromosome 17p11.2.
D. A centromere 17–specific FISH probe.
Rationale: Centromeric probes are used to detect the number of copies of a chromosome. Microdeletions cannot be detected by centromeric probes.
E. A subtelomeric probe for the short arm of chromosome 17.
Rationale: A subtelomeric probe cannot be used because the deletion in the SMS is in the proximal short arm. A subtelomeric probe will be useful only for distal deletion or duplication of a chromosome.
B. The karyotype shows an unbalanced translocation between the short arms of chromosomes 15 and 17, which results in partial trisomy of 15p and partial monosomy of 17p.
C. The karyotype shows an unbalanced translocation between the short arms of chromosomes 15 and 17, which results in partial monosomy of 15p and partial trisomy of 17p.
D. The karyotype shows an unbalanced derivative chromosome 17 resulting from a translocated insertion of short arm material from chromosome 15p at band p13.3 into 17p11.2.
E. The karyotype shows an unbalanced derivative chromosome 17 resulting from a translocated insertion of short arm material from chromosome 15p at band p11.2 into 17p13.3.
Rationale: The karyotype does not reflect an insertion but rather an unbalanced translocation between the short arms of chromosomes 15 and 17 (the imbalance results in partial trisomy of 15p and partial monosomy of 17p.) The abnormal chromosome is a derivative chromosome 17 that has extra material derived from the short arm of chromosome 15 (pter-15p11.2), which replaces the material deleted on chromosome 17 (17pter-p13.3).
Rationale: Sotos syndrome is caused by (1) a heterozygous mutation in the NSD1 gene or (2) a deletion in the 5q35 region including genomic sequence in addition to the NSD1 gene. The principal clinical features include overgrowth from the prenatal stage through childhood, distinctive facial features, large head circumference, pointed chin, and mild-to-severe learning difficulties.
B. SMS.
Rationale: SMS is caused in most cases (90%) by a 3.7-Mb interstitial deletion in the 17p11.2 region. This region is closer (i.e., more proximal) to the centromere than the translocation described in the current case. The disorder can also be caused by mutations in the RAI1 gene, which is within the Smith-Magenis 17p11.2 chromosome region. The principal clinical features include mild-to-severe learning disabilities, brachycephaly, midface hypoplasia, prognathism, hoarse voice, speech delay with or without hearing loss, psychomotor and growth retardation, and behavioral problems.
C. Cri-du-chat syndrome.
Rationale: Cri-du-chat syndrome is caused by a deletion of the short arm of chromosome 5 and is characterized by a high-pitched, monotonous cry; microcephaly; a round face; hypertelorism; epicanthic folds; micrognathia; impaired growth; severe developmental delay; and learning disability.
D. DiGeorge syndrome.
Rationale: DiGeorge syndrome is caused by a 1.5- to 3.0-Mb hemizygous deletion of the proximal long arm region of chromosome at 22q11.2. Haploinsufficiency of the TBX1 gene is believed to be responsible for most of the physical malformations. The principal clinical features include hypocalcemia arising from parathyroid hypoplasia, thymic hypoplasia, and cardiac outflow tract defects.
E. Miller-Dieker syndrome.
Rationale: Miller-Dieker syndrome is a contiguous gene deletion syndrome involving genes on chromosome 17p13.3. Deletions or mutations in the LIS1 gene appear to cause the lissencephaly. The principal clinical features include lissencephaly, cardiac malformations, microcephaly, wrinkled skin over the glabella and frontal suture, prominent occiput, narrow forehead, downward-slanting palpebral fissures, small nose and chin, hypoplastic male external genitalia, growth retardation, and mental deficiency with seizures and electroencephalographic abnormalities. A reduced lifespan is commonly observed.
Rationale: The karyotype is unbalanced with partial trisomy (extra genetic material) of the short arm of chromosome 15 and partial monosomy (missing genetic material) of the short arm of chromosome 17.
B. The missing genetic material from the short arm of chromosome 15 and the extra genetic material from the short arm of chromosome 17.
Rationale: The missing genetic material derives from the short arm of chromosome 17 and the extra genetic material from the short arm of chromosome 15.
C. The extra genetic material from the short arm of chromosome 15 and the missing genetic material from the short arm of chromosome 17.
Rationale: Although this statement reflects the genomic imbalance consistent with this karyotype, the associated phenotype is unlikely to be linked to the extra material from the short arm of chromosome 15. This is because the short arm of chromosome 15 (as well as the short arms of all the acrocentric chromosomes) contains highly variable amounts of ribosomal gene repeats; loss or gain of this genetic material is clinically insignificant because there are sufficient ribosomal genes on the remaining acrocentric short arms. The sizes of the short arms of the acrocentric chromosomes (and their resulting gene content) are highly polymorphic, with some individuals showing almost no visible short arm material and others showing extremely large short arms.
D. Only the missing genetic material from the short arm of chromosome 17.
Rationale: A deletion of the terminal region of chromosome 17 at 17p13.3 (including the LIS1 gene) is associated with a clinical diagnosis of the Miller-Dieker syndrome, which is characterized by lissencephaly with dysmorphic features. The extra material from the short arm of chromosome 15 contains multiple ribosomal gene repeats that already exist as a polymorphic entity; thus, this is unlikely to be a contributing factor to the clinical phenotype.
E. Only the extra genetic material from the short arm of chromosome 15.
Rationale: The short arm of chromosome 15 (as well as the short arms of all the acrocentric chromosomes) contains highly variable amounts of ribosomal gene repeats; gain (or loss) of this genetic material is clinically insignificant because there are sufficient ribosomal genes on the remaining acrocentric short arms. The size of the short arms of the acrocentric chromosomes (and their resulting gene content) is highly polymorphic, with some individuals showing almost no visible short arm material and others showing extremely large short arms. In this karyotype, the gain of the short arm material is unlikely to be pathogenic and the observed phenotype is likely due to the missing material on the short arm of chromosome 17.
Rationale: t(8;14)(q24;q32), t(8;22)(q24;q11), and t(2;8)(p11;q24) are the characteristic chromosome translocations in BL.
B. MZL.
Rationale: Chromosome alterations involving gains of the long arms of chromosomes 3 and 18 are commonly seen in MZLs.
C. Multiple myeloma, possibly with good prognosis.
D. Multiple myeloma, possibly with poor prognosis.
Rationale: 14q32 rearrangements are seen in approximately 50% of plasma cell neoplasia cases. The t(14;20)(q32;q11.2) abnormality is characteristic of multiple myeloma, and the presence of 1q duplications is indicative of a poor prognosis.
E. MALT lymphoma.
Rationale: The chromosome change of t(11;18)(q21;q21) is primarily detected in MALT lymphomas.
Rationale: The t(2;8)(p11.2;q24) translocation, resulting in MYC rearrangement with the IGK locus, is seen in BL and DLBCL. A complex karyotype associated with t(2;8) is particularly seen in DLBCL and BL.
B. MZL.
Rationale: The three distinct clinicopathologic subtypes of MZL exhibit no diagnostic chromosome changes. However, trisomies 3 and 18, 7q deletion, and 14q32 rearrangements with various partners are frequently seen.
C. Multiple myeloma.
Rationale: In multiple myeloma, the 14q32 region involving the IGH gene is frequently partnered with 11q13 (the cyclin D1 gene), 4p16 (the FGFR3 gene), 6p25 (the cyclin D3 gene), 16q23 (the MAF gene), and 20q11 (the MAFB gene), thereby forming balanced reciprocal translocations. Other changes frequently seen in multiple myeloma are deletions at 11q23 and 6q, 1q duplications, and trisomies of chromosomes 3, 5, 7, 9, 11, 15, 19, and 21.
D. MALT lymphoma.
Rationale: The t(11;18)(q21;q21) balanced translocation is primarily detected in MALT lymphoma.
E. HL.
Rationale: HL is not associated with t(2;8). Specific chromosome changes have not been reported for HL. However, chromosome deletions involving 1p, 6q, 7q, and duplications of 1q have been seen.
Rationale: This karyotype indicates deletion 5q. Deletion of chromosome 5q as a sole chromosome abnormality is also known as the 5q– syndrome and represents a distinct clinical entity. It presents with a macrocytic anemia and a normal or elevated platelet count. Abnormalities in the megakaryocytic lineage are prominent. This deletion is associated with a favorable outcome with low rates of leukemic transformation and a relatively long survival (i.e., several years).
B. 47,XY,+8.
Rationale: This karyotype indicates trisomy 8. This abnormality is seen in all MDS subgroups, both as an isolated chromosome abnormality and with other recurring chromosome abnormalities known to have prognostic significance. This abnormality is associated with a variable clinical course and is expected to be associated with intermediate prognosis.
C. 46,XY,del(20)(q11.2q13.3).
Rationale: This karyotype indicates deletion of the long arm of chromosome 20. Patients with deletion 20q have prominent dysplasia in the erythroid and megakaryocytic lineages. These patients have low disease risk (usually refractory anemia), a low rate of progression to acute leukemia, and prolonged survival (i.e., a median of 45 months versus 28 months for other MDS patients).
D. 45,X,-Y.
Rationale: This karyotype indicates loss of the Y chromosome. Loss of the Y chromosome as a sole cytogenetic abnormality in a patient with a diagnosis of MDS (clinical and bone marrow morphology findings) is associated with a favorable outcome.
E. 46,XY,del(7)(q11.2q36).
Rationale: This karyotype indicates deletion 7q. Deletion 7q or monosomy 7 is associated with poor outcome in terms of both survival and progression to acute leukemia.
Rationale: Gain of chromosome 7 and loss of chromosome 10 are common genetic changes in astrocytomas.
B. Medulloblastomas.
Rationale: An isochromosome 17q in brain tumors is pathognomonic for medulloblastoma.
C. Meningiomas.
Rationale: Loss of chromosome 22 is the most common cytogenetic aberration in meningiomas.
D. Oligodendrogliomas.
Rationale: Patients whose oligodendrogliomas have concomitant 1p and 19q deletions have a significantly longer survival than those without deletions.
E. Ependymomas.
Rationale: Deletions of chromosome 19 and 22 are recurring chromosome abnormalities in ependymomas.
Rationale: Chromophobe RCCs exhibit a unique combination of chromosome losses with a modal chromosome number of 38-39. Monosomy of chromosomes 1, 2, 6, 10, 13, 17, and 21 are most frequent.
B. Renal oncocytoma.
Rationale: Loss of chromosomes 1 and 14, and the Y chromosome, along with 11q13 rearrangements are characteristic of a renal oncocytoma.
C. Papillary RCC.
Rationale: A combination of trisomy or tetrasomy of chromosome 7, trisomy 17, and loss of the Y chromosome is a frequent feature of papillary RCC. Additional trisomies of chromosomes 12, 16, and 20 are seen in aggressive tumors.
D. Conventional clear cell RCC.
Rationale: Chromosome 3p deletions involving the 3p21 region are a characteristic feature of this neoplasm.
E. Renal angiomyolipoma.
Rationale: Chromosome abnormalities are rare in renal angiomyolipomas. Trisomy 7 was reported in a few cases.
Rationale: Embryonal rhabdomyosarcomas do not show any recurrent balanced chromosome translocations.
B. Primitive neuroectoderal tumor.
Rationale: t(11;22)(q24;q12), resulting in fusion of the EWS gene on 22q12 with FLI1 at 11q24, is a classic feature of primitive neuroectoderal tumor/Ewing sarcoma and serves as a diagnostic chromosome translocation for Ewing sarcoma.
C. Giant cell tumor of bone.
Rationale: Giant cell tumor of bone shows a near-diploid range chromosomes; no diagnostic cytogenetic change has yet been identified.
D. Lipoma.
Rationale: Rearrangements of 12q13-15 regions are seen in two thirds of clonally abnormal cases, with 3q27-29 as the most common translocation partner.
E. Alveolar rhabdomyosarcoma.
Rationale: Most alveolar rhabdomyosarcoma cases exhibit characteristic chromosome translocations, such as t(2;13)(q36;q14) or t(1;13)(p36;q14). The FOXO1 gene on 13q14 rearranges with the PAX3 gene on 2q35 and the PAX7 gene on 1p36 and results in formation of fusion gene products.
Rationale: The hallmark of CML is the t(9;22)(q34;q11.2) translocation and its associated variant translocations. A high hyperdiploid karyotype is not a common feature of CML.
B. MDS, predicting a good prognosis.
Rationale: The chromosome changes frequently seen in MDS are del(5q)/monosomy 5, del(7q)/monosomy 7, del(20q)/monosomy 20, and trisomy 8. A high hyperdiploid karyotype is not a feature of MDS.
C. Pre-B-cell ALL (pre-B ALL), predicting a poor prognosis.
D. Pre-B ALL, predicting a good prognosis.
Rationale: The described immunophenotype is characteristic of pre-B ALL and a high hyperdiploid karyotype is commonly seen in a subset of pre-B ALL patients. A karyotype showing hyperdiploidy is associated with a high sensitivity to chemotherapy and a better clinical outcome, thus predicting a good prognosis.
E. AML, predicting a good prognosis.
Rationale: AML is associated with specific chromosome translocations, such as t(15;17)(q22;q21), t(16;16)(p13;q22), t(8;21)(q22;q22), and other loss or gain of chromosome regions. A high hyperdiploid karyotype is not commonly seen in AML.
Rationale: Common chromosome changes associated with MDS are deletion 5q/monosomy 5, deletion 7q/monosomy 7, deletion 20q/monosomy 20, and trisomy 8.
B. The t(15;17)(q22;q21) translocation is diagnostic of CML.
Rationale: t(15;17)(q21;q22) is not associated with CML. CML is characterized by t(9;22)(q34;q11.2).
C. The t(15;17)(q22;q21) translocation is diagnostic of APL but does not predict treatment outcome.
Rationale: The t(15;17)(q22;q21) translocation is diagnostic of APL. Because these lesions exhibit high sensitivity to treatment, this predicts a good prognosis.
D. The chromosomal translocation (15;17)(q22;q21) is not associated with acute myelocytic leukemia.
Rationale: The t(15;17)(q22;q21) translocation is specific to the FAB M3 subtype of acute myelocytic leukemia and is pathognomonic of APL.
E. The t(15;17)(q22;q21) translocation is diagnostic of APL and predicts a favorable treatment outcome.
Rationale: t(15;17)(q22;q21) and the associated PML/RARA rearrangement is diagnostic of APL. These neoplastic cells are highly sensitive to treatment with ATRA, which acts as a differentiating agent. This translocation predicts a good prognosis in this disorder.
Rationale: Chromophobe RCCs exhibit a unique combination of chromosome losses with a modal chromosome number of 38-39. Monosomy of chromosomes 1, 2, 6, 10, 13, 17, and 21 are most frequent.
B. Renal oncocytoma.
Rationale: Loss of chromosomes 1 and 14 and the Y chromosome, along with 11q13 rearrangements, are characteristic of a renal oncocytoma.
C. Papillary RCC.
Rationale: A combination of trisomy or tetrasomy of chromosome 7, trisomy 17, and loss of the Y chromosome is a frequent feature of papillary RCC. Additional trisomies of chromosomes 12, 16, and 20 are seen in aggressive tumors.
D. Conventional clear cell RCC.
Rationale: Chromosome 3p deletions involving the 3p21 region are a characteristic feature of this neoplasm.
E. Renal angiomyolipoma.
Rationale: Chromosome abnormalities are rare in renal angiomyolipomas. Trisomy 7 was reported in a few cases.
Rationale: Deletion of chromosome 3p is a characteristic finding in clear cell RCC.
B. Papillary RCC.
Rationale: Combinations of trisomy 7 and 17 are commonly found in papillary RCC.
C. Chromophobe RCC.
Rationale: Combination monosomies (chromosomes 1, 2, 6, 10, 13, 17 and/or 21) are found in chromphobe RCC.
D. Mucinous tubular and spindle cell carcinoma.
Rationale: Loss of chromosomes 1, 14, and 15 are found in these tumors.
E. Oncocytoma.
Rationale: Renal oncotyoma is a benign neoplasm. Chromosome abnormalities are seen in approximately 50% of oncocytomas. Loss of chromosomes 1 and 14, and loss of the Y chromosome in males, are seen in one subset of tumors. In a second subset, translocations at 11q13 are seen. 11q13 rearranges with many different chromosomes in this setting.
Rationale: Although the correct chromosomes are shown here, the letters “der” should be used instead of “t” and the translocation breakpoint should be (q10;q10).
B. 45,XX,der(13;21)(q10;q10).
C. 46,XX,t(13;21)(p10;q10).
D. 46,XX,der(13),t(13;21)(p10;p10).
Rationale: The Robertsonian derivative always involves both chromosomes; that is, der(13;21).
E. 46,XX,der(13;21)(q10;p10).
Rationale for B, C, and E: Balanced carriers of a Robertsonian translocation have 45 chromosomes. The translocation is described as a derivative chromosome and thus the use of “der” is appropriate. The breakpoints for Robertsonian translocations are always at (q10;q10).
Rationale: The t(X;18) is found in almost all synovial sarcomas; this translocation results in either the SS18-SSX1, SS18-SSX2, or SS18-SSX4 fusion genes.
B. Desmoplastic small round cell tumor.
Rationale: The characteristic translocation in this tumor is t(11;22)(p13;q12), resulting in EWSR1-WT1 fusion transcript.
C. Embryonal rhabdomyosarcoma.
Rationale: These tumors have complex karyotypes, but no recurrent translocations have been identified.
D. Alveolar rhabdomyosarcoma.
Rationale: The cytogenetic hallmark of alveolar rhabdomyosarcomas is t(2;13)(q36;q14) and, less frequently, t(1;13)(p36;q14); these translocations result in PAX3-FOXO1A and PAX7-FOXO1A fusions, respectively.
E. Myxoid liposarcoma.
Rationale: These tumors have a t(12;16)(q13;p11), which results in a FUS-DDIT3 chimeric gene.
Rationale: Embryonal rhabdomyosarcomas do not show any recurrent balanced chromosome translocations.
B. Alveolar rhabdomyosarcoma.
Rationale: Alveolar rhabdomyosarcomas exhibit the following characteristic balanced chromosome translocations: t(2;13)(q36;q14) or t(1;13)(p36;q14). The FOXO1 gene on 13q14 rearranges with the PAX3 gene on 2q35 and the PAX7 gene on 1p36, resulting in the formation of fusion gene products.
C. Primitive neuroectoderal tumor.
Rationale: t(11;22)(q24;q12), resulting in the fusion of the EWS gene on 22q12 with FLI1 at 11q24, is a classic feature of a primitive neuroectoderal tumor/Ewing sarcoma and serves as diagnostic marker for this neoplasm.
D. Synovial sarcoma.
Rationale: The characteristic chromosome translocation in synovial sarcoma is the recombination between the Xp11.2 and 18p11.2 regions, resulting in t(X;18)(p11.2;q11.2). This translocation results in fusion genes between SS18 (SYT) on 18p with one of three genes (i.e., SSX1, SSX2, or SSX4) in the Xp11.2 region.
E. Myxoid liposarcoma.
Rationale: The cytogenetic hallmark of a myxoid liposarcoma is a balanced translocation, t(12;16)(q13;p11.2), resulting in formation of the FUS-DDIT3 fusion gene.
Rationale: The t(8;14)(q24;q32) translocation, resulting in rearrangement of the MYC gene with the IGH locus, is the characteristic chromosome translocation in BL.
B. ALCL.
Rationale: A balanced translocation between chromosome regions 2p23 and 5q35 [t(2;5)(p23;q35)], resulting in ALK rearrangement, is a classic feature of ALCL.
C. Multiple myeloma.
Rationale: In multiple myeloma, the 14q32 region involving the IGH gene is frequently partnered with 11q13 (the cyclin D1 gene), 4p16 (the FGFR3 gene), 6p25 (the cyclin D3 gene), 16q23 (the MAF gene), and 20q11 (the MAFB gene), thereby forming balanced reciprocal translocations.
D. T-PLL.
Rationale: The immunophenotype in the current case is consistent with a mature T-cell leukemia/lymphoma. The inv(14)(q11.2q32) cytogenetic abnormality is frequently seen (~ 65% of cases) in T-cell prolymphocytic leukemia.
E. HL.
Rationale: HL is not associated with inv(14). Specific chromosome changes have not been reported in HL. However, chromosome deletions involving 1p, 6q, 7q, and duplications of 1q, can be seen.
Rationale: This karyotype shows an extra copy of chromosome 18, which is associated with a clinical diagnosis of trisomy 18. Trisomy 18 is characterized by multiple congenital abnormalities and developmental delay.
B. 46,XX,t(7;18)(q21;q21).
Rationale: This karyotype shows a balanced translocation between the long arms of chromosomes 7 and 18. Balanced translocations are not typically associated with multiple congenital abnormalities and dysmorphic features unless (1) there is an accompanying submicroscopic microdeletion/microduplication at the breakpoint regions, or (2) the breakpoint occurs in a critical region with concomitant disruption of the gene function.
C. 47,XX,+16.
Rationale: This karyotype shows an extra copy of chromosome 16 (trisomy 16), which is always lethal in the first or second trimester.
D. 47,XXX.
Rationale: This karyotype shows an extra copy of the X chromosome, which is associated with a clinical diagnosis of the triple X syndrome. Physical development in females with XXX is generally unremarkable and there are no phenotypic abnormalities. Some may be tall, be at risk for learning disabilities, and may have fertility problems. There is a general tendency for IQ to be slightly reduced compared with siblings.
E. 46,XX,inv(9)(p12q13).
Rationale: This karyotype shows a balanced pericentric inversion of chromosome 9. Most pericentric inversions confer an increased risk for miscarriages and offspring with congenital abnormalities and/or mental retardation as a result of unbalanced segregation during gametogenesis. However, this particular inversion is a well-established benign polymorphism with no associated clinical risks.
Rationale: Risk of trisomy 21 or Down syndrome increases with advanced maternal age. Age-associated risk for trisomy 21 at 16 weeks (amniocentesis) is 1:620 for a 30-year-old woman.
B. A woman with the karyotype 45,XX,t(21;21)(q10;q10).
Rationale: A woman with this karyotype can only have trisomy 21 or monosomy 21 conceptions.
C. A woman with no family history of Down syndrome.
Rationale: A woman with no family history may still have an age-associated risk for Down syndrome.
D. A woman with the karyotype 45,XX,t(14;21)(q10;q10).
Rationale: Female carriers of this translocation have an approximately 15% risk of trisomy 21. Nonetheless, the woman described in Case B is at a greater risk for a trisomy 21 live birth.
E. A 35-year-old woman who had a previous child with trisomy 21.
Rationale: Age-associated risk for trisomy 21 at 16 weeks (i.e., at amniocentesis) is 1:245 for a 35-year-old woman.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


