Warfarin-induced skin necrosis
Heparin-induced skin necrosis
Heparin delayed-type hypersensitivity
Incidence
0.01–0.1 %
UFH: <3 %
LMWH: <0.1 %
7.5 %
Onset
90 % day 3–6
Almost all by day 10
Day 5–10
If previous sensitization (within 3 months), day 1–5
Day 7–14
If previous sensitization (within 100 days), day 1–5
Gross appearance
Necrosis at sites with increased subcutaneous fat
Necrosis at injection sites but can be at distant sites. Can be caused by IV heparin.
Erythematous plaques at injection sites but can be at distant sites. Can be caused by IV heparin.
Histopathology
Fibrin thrombi (red clots) in dermal vessels
Necrosis
RBC extravasation
Platelet thrombi (white clots) in dermal vessels
Necrosis
RBC extravasation
Perivascular lymphocytic infiltrate
± Spongiosis
Associated features
Protein C deficiency
Obesity
Female sex
VTE
Pain
HITT
Pruritis
Pregnancy
Course
Self-limiting
Life-threatening
With continued course, new and extensive skin necrosis
Self-limiting
With continued course, may generalize
Management
Discontinue warfarin
Start vitamin K, FFP
Start alternative anticoagulation
Discontinue heparin
Start heparinoid or direct thrombin inhibitor
Discontinue heparin
± Allergy testing
Ability to restart
Yes (at low dose and slow taper)
No (with exception of specific surgical situations)
No SQ UFH or LMWH.
IV heparin and fondaparinux often tolerated
Warfarin-Induced Skin Necrosis (WISN)
Warfarin is one of the coumarin congeners, which also include bishydroxycoumarin, phenprocoumon, acenocoumarol. These medications anticoagulate by inhibiting the enzyme that reduces oxidized vitamin K back to its active state, thereby inhibiting vitamin K-dependent coagulation factors. Warfarin is the most widely used oral anticoagulant worldwide.
Epidemiology and Pathophysiology
WISN affects 0.01–0.1 % of treated patients. It occurs more frequently in obese middle-aged women, with a female-to-male ratio of 4:1. The majority of patients are ill and hospitalized; DVT, pulmonary embolism, and thrombophlebitis are the most common indications for anticoagulation in these patients.
The pathophysiology of WISN involves the balance of anticoagulation and coagulation forces, perturbed by the initiation of warfarin. Factors inhibited by warfarin, due to their vitamin K-dependence, include factors II, VII, IX, and X as well as anticoagulation proteins C and S. Protein C and factor VII have short half-lives (5–8 h) relative to factors II, IX, and X (2–3 days), leading to a more rapid decrease in the former relative to the latter. This causes a transient hypercoagulable state during the initiation of therapy. This also explains why protein C deficiency (acquired or inherited) is a significant risk factor for development of WISN. Less frequently, protein S deficiency, antithrombin III deficiency, factor V Leiden mutation, and antiphospholipid antibody syndrome have been associated with WISN. Other proposed mechanisms of WISN include the direct toxic effect of warfarin on vessel walls and immunologic hypersensitivity to warfarin.
Presentation
Early symptoms are localized paresthesia and edema with progression to livedo macules, petechiae, and ecchymosis; hemorrhagic bullae form within 24 h (Fig. 27.1). After days, lesions are characterized by full-thickness necrosis and painful subcutaneous ulcerations. 90 % of cases occur between days 3 and 6 after treatment initiation; almost all occur by day 10. There have been reports of WISN occurring up to 15 years after treatment initiation as well as several days after cessation of therapy. Areas with more subcutaneous fat are more susceptible, such as the abdomen, buttocks, thighs, and breast tissue. One-third of patients have multiple sites of involvement. Of note, the cutaneous appearance of warfarin necrosis is difficult to distinguish from that of heparin necrosis.
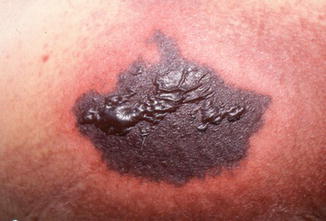
Fig. 27.1
Necrotic tissue measuring 15 cm over the left buttock of a patient on day 3 of coumadin therapy. At least 90 % of patients are on days 3–5 of coumadin therapy. Bullae formation is seen over the surface and dramatic surrounding erythema. Pain is usually severe
Histopathology
Diffuse microthrombi in dermal and subcutaneous vessels
Necrosis of epidermis and dermis
Erythrocyte extravasation
No evidence of inflammation or vasculitis
Differential Diagnosis
HISN: history of heparin use, lesion at injection site, heparin-induced thrombocytopenia and thrombosis syndrome (HITT) manifestations
Catastrophic antiphospholipid syndrome (Asherson’s syndrome): multiorgan dysfunction, positive antiphospholipid antibodies, histopathologic evidence of small vessel thromboses
Calciphylaxis (Fig. 27.2): end-stage renal disease, characteristic histopathology, predominant lower extremity involvement
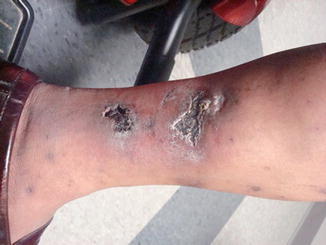
Fig. 27.2
Multiple sites of necrosis over the ankle of a dialysis patient with surrounding erythema and induration. Histopathology on biopsy was compatible with calciphylaxis. The pain was exquisite and response to sodium thiosulfate was rapid. There was no history of anticoagulation
Microemboli (septic, cholesterol): “purple toe syndrome”
Disseminated intravascular coagulation (Fig. 27.3—symmetrical peripheral gangrene and purpura fulminans are both probably the same disease): associated clinical condition (sepsis, hemolytic transfusion reaction, severe head injury, amniotic or fat embolism, etc.), multiple organ ischemic necrosis, consistent labs (schistocytes on blood smear, thrombocytopenia, prolonged coagulation studies, increased D-dimer and fibrin degradation products, reduced coagulation factor levels)
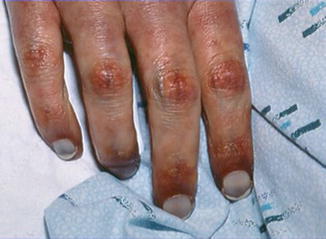
Fig. 27.3
Purpuric necrotic changes in fingers in a patient with disseminated intravascular coagulation syndrome. In purpura fulminans the necrosis is often symmetric and peripheral. The patient had E coli sepsis. Sepsis is the commonest cause of purpura fulminans
Purpura fulminans (Fig. 27.3): diffuse nonthrombocytopenic purpura, recent serious infection
Necrotizing fasciitis: positive wound culture and tissue Gram stain
Cryoglobulinemia: palpable purpura, Raynaud phenomenon, elevated cryocrit, leukocytoclastic vasculitis, hepatitis C, decreased C4 out of proportion to C3
Diagnosis and Management
Diagnosis is made based on clinical suspicion, biopsy, and ruling out other diagnoses. First steps in management include cessation of warfarin, administration of fresh frozen plasma and vitamin K, and initiation of an alternative anticoagulant such as heparin. Local treatment includes topical bactericidal agents. Surgical debridement, skin grafting, or amputation is required in more than 50 % of cases. Treatment with recombinant protein C has been shown to be beneficial in patients with documented protein C deficiency, but cost is prohibitive. An important distinction between warfarin-induced and heparin-induced skin necrosis is that warfarin can be reintroduced after an episode while heparin cannot, in most circumstances. It is important to start warfarin at a low dose, with slow increase to therapeutic level; loading doses should be avoided, and heparin bridging considered. The lesions are self-limited and generally resolve over several weeks.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


