CHAPTER 34 Control of Body Fluid Osmolality and Volume
The kidneys maintain the osmolality and volume of body fluids within a narrow range by regulating the excretion of water and NaCl, respectively. This chapter discusses the regulation of renal water excretion (urine concentration and dilution) and NaCl excretion. The composition and volumes of the various body fluid compartments are reviewed in Chapter 2.
CONTROL OF BODY FLUID OSMOLALITY: URINE CONCENTRATION AND DILUTION
As described in Chapter 2, water constitutes approximately 60% of the healthy adult human body. Body water is divided into two compartments (i.e., intracellular fluid [ICF] and extracellular fluid [ECF]), which are in osmotic equilibrium. Water intake into the body generally occurs orally. However, in clinical situations, intravenous infusion is an important route of water entry.
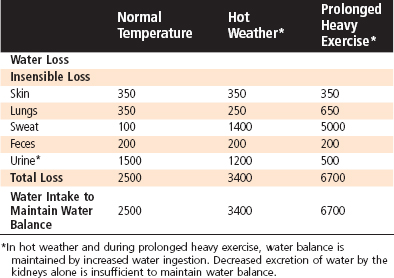
The kidneys are responsible for regulating water balance and under most conditions are the major route for elimination of water from the body (Table 34-1). Other routes of water loss from the body include evaporation from cells of the skin and respiratory passages. Collectively, water loss by these routes is termed insensible water loss because the individual is unaware of its occurrence. The production of sweat accounts for the loss of additional water. Water loss by this mechanism can increase dramatically in a hot environment, with exercise, or in the presence of fever (Table 34-2). Finally, water can be lost from the gastrointestinal tract. Fecal water loss is normally small (≈100 mL/day) but can increase dramatically with diarrhea (e.g., 20 L/day with cholera). Vomiting can also cause gastrointestinal water loss.
Table 34-1 Normal Routes of Water Gain and Loss in Adults at Room Temperature (23° C)
| Route | mL/Day |
|---|---|
| Water Intake | |
| Fluid* | 1200 |
| In food | 1000 |
| Metabolically produced from food | 300 |
| TOTAL | 2500 |
| Water Output | |
| Insensible | 700 |
| Sweat | 100 |
| Feces | 200 |
| Urine | 1500 |
| TOTAL | 2500 |
* Fluid intake varies widely for both social and cultural reasons.
Table 34-2 Effect of Environmental Temperature and Exercise on Water Loss and Intake (mL/day) in Adults
The following sections discuss the mechanisms by which the kidneys excrete either hypoosmotic (dilute) or hyperosmotic (concentrated) urine. The control of vasopressin secretion and its important role in regulating excretion of water by the kidneys are also explained (see also Chapter 40).
Antidiuretic Hormone
Antidiuretic hormone (ADH), or vasopressin, acts on the kidneys to regulate the volume and osmolality of urine. When plasma ADH levels are low, a large volume of urine is excreted (diuresis), and the urine is dilute.* When plasma levels are high, a small volume of urine is excreted (antidiuresis), and the urine is concentrated.
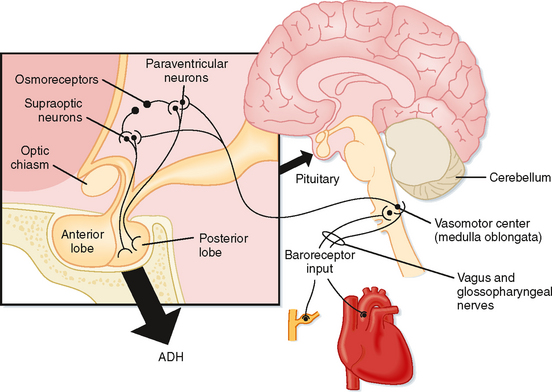
ADH is a small peptide that is nine amino acids in length. It is synthesized in neuroendocrine cells located within the supraoptic and paraventricular nuclei of the hypothalamus.* The synthesized hormone is packaged in granules that are transported down the axon of the cell and stored in nerve terminals located in the neurohypophysis (posterior pituitary). The anatomy of the hypothalamus and pituitary gland is shown in Figure 34-1.
Osmotic Control of ADH Secretion
Changes in the osmolality of body fluids play the most important role in regulating secretion of ADH; changes as minor as 1% are sufficient to alter it significantly. Although neurons in the supraoptic and paraventricular nuclei respond to changes in body fluid osmolality by altering their secretion of ADH, it is clear that there are separate cells in the anterior hypothalamus that are exquisitely sensitive to changes in body fluid osmolality and therefore play an important role in regulating the secretion of ADH.* These cells, termed osmoreceptors, appear to behave as osmometers and sense changes in body fluid osmolality by either shrinking or swelling. The osmoreceptors respond only to solutes in plasma that are effective osmoles (see Chapter 1). For example, urea is an ineffective osmole when the function of osmoreceptors is considered. Thus, elevation of the plasma urea concentration alone has little effect on ADH secretion.
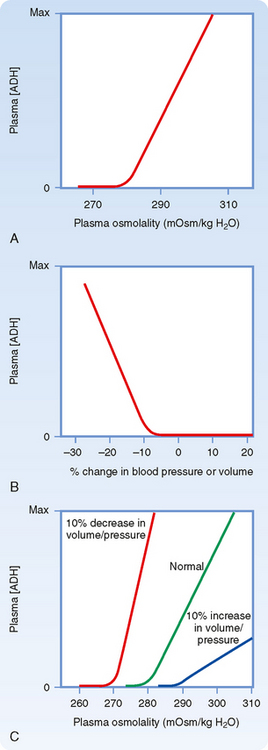
Figure 34-2, A, illustrates the effect of changes in plasma osmolality on circulating ADH levels. The slope of the relationship is quite steep and accounts for the sensitivity of this system. The set point of the system is the plasma osmolality value at which ADH secretion begins to increase. Below this set point, virtually no ADH is released. The set point varies among individuals and is genetically determined. In healthy adults, it varies from 275 to 290 mOsm/kg H2O (average, ≈280 to 285 mOsm/kg H2O). Several physiological factors can also change the set point in a given individual. As discussed later, alterations in blood volume and pressure can shift it. In addition, pregnancy is associated with a decrease in the set point.
Hemodynamic Control of ADH Secretion
A decrease in blood volume or pressure also stimulates secretion of ADH. The receptors responsible for this response are located in both the low-pressure (left atrium and large pulmonary vessels) and the high-pressure (aortic arch and carotid sinus) sides of the circulatory system. Because the low-pressure receptors are located in the high-compliance side of the circulatory system (i.e., venous) and because the majority of blood is in the venous side of the circulatory system, these low-pressure receptors can be viewed as responding to overall vascular volume. The high-pressure receptors respond to arterial pressure. Both groups of receptors are sensitive to stretch of the wall of the structure in which they are located (e.g., cardiac atrial wall, wall of the aortic arch) and are termed baroreceptors. Signals from these receptors are carried in afferent fibers of the vagus and glossopharyngeal nerves to the brainstem (solitary tract nucleus of the medulla oblongata), which is part of the center that regulates heart rate and blood pressure (see also Chapter 18). Signals are then relayed from the brainstem to the ADH-secreting cells of the supraoptic and paraventricular hypothalamic nuclei. The sensitivity of the baroreceptor system is less than that of the osmoreceptors, and a 5% to 10% decrease in blood volume or pressure is required before ADH secretion is stimulated. This is illustrated in Figure 34-2, B. A number of substances have been shown to alter the secretion of ADH through their effects on blood pressure, including bradykinin and histamine, which lower pressure and thus stimulate ADH secretion, and norepinephrine, which increases blood pressure and inhibits ADH secretion.
Alterations in blood volume and pressure also affect the response to changes in body fluid osmolality (see Fig. 34-2, C). With a decrease in blood volume or pressure, the set point is shifted to lower osmolality values and the slope of the relationship is steeper. In terms of survival of the individual, this means that when faced with circulatory collapse, the kidneys will continue to conserve water, even though by doing so they reduce the osmolality of body fluids. With an increase in blood volume or pressure, the opposite occurs. The set point is shifted to higher osmolality values, and the slope is decreased.
Actions of ADH on the Kidneys
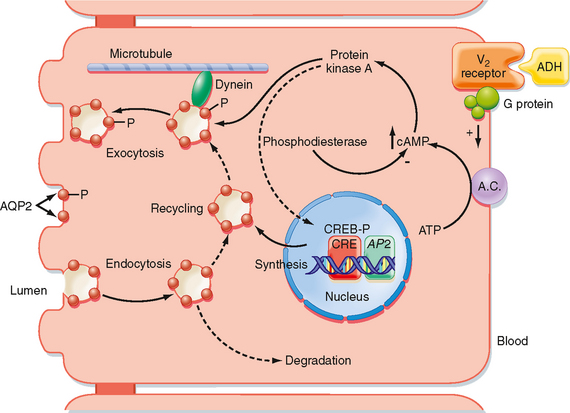
The actions of ADH on permeability of the collecting duct to water have been studied extensively. ADH binds to a receptor on the basolateral membrane of the cell. This receptor is termed the V2 receptor (i.e., vasopressin 2 receptor).* Binding to this receptor, which is coupled to adenylyl cyclase via a stimulatory G protein (Gs), increases intracellular levels of cAMP. The rise in intracellular cAMP activates protein kinase A (PKA), which ultimately results in the insertion of vesicles containing aquaporin-2 (AQP2) water channels into the apical membrane of the cell, as well as the synthesis of more AQP2 (Fig. 34-3). With the removal of ADH, these water channels are reinternalized into the cell, and the apical membrane is once again impermeable to water. This shuttling of water channels into and out of the apical membrane provides a rapid mechanism for controlling permeability of the membrane to water. Because the basolateral membrane is freely permeable to water as a result of the presence of AQP3 and AQP4 water channels, any water that enters the cell through apical membrane water channels exits across the basolateral membrane, thereby resulting in net absorption of water from the tubule lumen.
In addition to the acute effects of ADH just described, ADH regulates the expression of AQP2 (and AQP3). When large volumes of water are ingested over an extended period (e.g., psychogenic polydipsia), expression of AQP2 and AQP3 in the collecting duct is reduced. As a consequence, when water ingestion is restricted, these individuals cannot maximally concentrate their urine. Conversely, in states of restricted water ingestion, expression of AQP2 and AQP3 in the collecting duct increases and thus facilitates the excretion of maximally concentrated urine.
The gene for the V2 receptor is located on the X chromosome. It codes for a 371–amino acid protein that is in the family of receptors that have seven membrane-spanning domains and are coupled to heterotrimeric G proteins. As shown in Figure 34-3, binding of ADH to its receptor on the basolateral membrane activates adenylyl cyclase. The increase in intracellular cAMP then activates protein kinase A (PKA), which results in phosphorylation of AQP2 water channels, as well as increased transcription of the AQP2 gene via activation of a cAMP-response element (CRE). Vesicles containing phosphorylated AQP2 move toward the apical membrane along microtubules driven by the molecular motor dynein. Once near the apical membrane, proteins called SNAREs interact with vesicles containing AQP2 and facilitate fusion of these vesicles with the membrane. The addition of AQP2 to the membrane allows water to enter the cell driven by the osmotic gradient (lumen osmolality < cell osmolality). The water then exits the cell across the basolateral membrane through AQP3 and AQP4 water channels, which are constitutively present in the basolateral membrane. When the V2 receptor is not occupied by ADH, the AQP2 water channels are removed from the apical membrane by clathrin-mediated endocytosis, thus rendering the apical membrane once again impermeable to water. The endocytosed AQP2 molecules may be either stored in cytoplasmic vesicles, ready for reinsertion into the apical membrane when ADH levels in plasma increase, or degraded.
ADH also increases the permeability of the terminal portion of the inner medullary collecting duct to urea. This results in an increase in reabsorption of urea and an increase in the osmolality of the medullary interstitial fluid. The apical membrane of medullary collecting duct cells contains two different urea transporters (UT-A1 and UT-A3).* ADH, acting through the cAMP/PKA cascade, increases permeability of the apical membrane to urea. This increase in permeability is associated with phosphorylation of UT-A1 and perhaps also UT-A3. Increasing the osmolality of the interstitial fluid of the renal medulla also increases the permeability of the collecting duct to urea. This effect is mediated by the phospholipase C pathway and involves phosphorylation by protein kinase C. Thus, this effect is separate and additive to that of ADH.
Thirst
The neural centers involved in regulating water intake (the thirst center) are located in the same region of the hypothalamus involved in regulating ADH secretion. However, it is not certain whether the same cells serve both functions. Indeed, the thirst response, like the regulation of ADH secretion, occurs only in response to effective osmoles (e.g., NaCl). Even less is known about the pathways involved in the thirst response to decreased blood volume or pressure, but it is believed that they are the same as those involved in the volume- and pressure-related regulation of ADH secretion. Angiotensin II, acting on cells of the thirst center (subfornical organ), also evokes the sensation of thirst. Because angiotensin II levels are increased when blood volume and pressure are reduced, this effect of angiotensin II contributes to the homeostatic response that restores and maintains body fluids at their normal volume.
The collecting ducts of some individuals do not respond normally to ADH. These individuals cannot maximally concentrate their urine and consequently have polyuria and polydipsia. This clinical entity is termed nephrogenic diabetes insipidus to distinguish it from central diabetes insipidus. Nephrogenic diabetes insipidus can result from a number of systemic disorders and, more rarely, occurs as a result of inherited disorders. Many of the acquired forms of nephrogenic diabetes insipidus are the result of decreased expression of AQP2 in the collecting duct. Decreased expression of AQP2 has been documented in the urine-concentrating defects associated with hypokalemia, lithium ingestion (some degree of nephrogenic diabetes insipidus develops in 35% of individuals who take lithium for bipolar disorder), ureteral obstruction, a low-protein diet, and hypercalcemia. The inherited forms of nephrogenic diabetes insipidus reflect mutations in the ADH receptor (V2 receptor) or the AQP2 molecule. Of these, approximately 90% of hereditary forms of nephrogenic diabetes insipidus are the result of mutations in the V2 receptor gene, with the other 10% being the result of mutations in the AQP2 gene. Because the gene for the V2 receptor is located on the X chromosome, these inherited forms are X-linked. To date, more than 150 different mutations in the V2 receptor gene have been described. Most of the mutations result in trapping of the receptor in the endoplasmic reticulum of the cell; only a few cases result in the surface expression of a V2 receptor that will not bind ADH. The gene coding for AQP2 is located on chromosome 12 and is inherited as both an autosomal recessive and an autosomal dominant defect. As noted in Chapter 1, aquaporins exist as homotetramers. This homotetramer formation explains the difference between the two forms of nephrogenic diabetes insipidus. In the recessive form, heterozygotes produce both normal AQP2 and defective AQP2 molecules. The defective AQP2 monomer is retained in the endoplasmic reticulum of the cell, and thus the homotetramers that do form contain only normal molecules. Accordingly, mutations in both alleles are required to produce nephrogenic diabetes insipidus. In the autosomal dominant form, the defective monomers can form tetramers with normal monomers, as well as with defective monomers. However, these tetramers are unable to traffic to the apical membrane.
Water intake is also influenced by social and cultural factors. Thus, individuals will ingest water even in the absence of the thirst sensation. Normally, the kidneys are able to excrete this excess water because they can excrete up to 18 L/day of urine. However, in some instances, the volume of water ingested exceeds the kidneys’ capacity to excrete water, especially over short periods. When this occurs, the body fluids become hypoosmotic. An example of how water intake can exceed the capacity of the kidneys to excrete water is long-distance running. A recent study of participants in the Boston Marathon found that hyponatremia developed in 13% of the runners during the course of the race.* This reflected the practice of some runners of ingesting water, or other hypotonic drinks, during the race to remain “well hydrated.” In addition, water is produced from the metabolism of glycogen and triglycerides used as fuels by the exercising muscle. Because over the course of the race they ingested, as well as generated through metabolism, more water than their kidneys were able to excrete or was lost by sweating, hyponatremia developed. In some racers the hyponatremia was severe enough to elicit the neurological symptoms described previously.
One can find throughout the popular press the admonition to drink eight 8-oz glasses of water a day (the 8 × 8 recommendation). Drinking this volume of water is said to provide innumerable health benefits. As a result, it seems that everyone now has a water bottle as a constant companion. Although ingesting this volume of water over the course of a day (approximately 2 L) will not harm most individuals, there is no scientific evidence to support the beneficial health claims ascribed to the 8 × 8 recommendation.† Indeed most individuals get adequate amounts of water through the food that they ingest and the fluids taken with those meals.
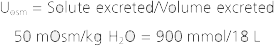
Renal Mechanisms for Dilution and Concentration of Urine
Under normal circumstances, excretion of water is regulated separately from excretion of solutes. For this to occur, the kidneys must be able to excrete urine that is either hypoosmotic or hyperosmotic with respect to body fluids. This ability to excrete urine of varying osmolality in turn requires that solute be separated from water at some point along the nephron. As discussed in Chapter 33, reabsorption of solute in the proximal tubule results in reabsorption of a proportional amount of water. Hence, solute and water are not separated in this portion of the nephron. Moreover, this proportionality between proximal tubule water and solute reabsorption occurs regardless of whether the kidneys excrete dilute or concentrated urine. Thus, the proximal tubule reabsorbs a large portion of the filtered load of solute and water, but it does not produce dilute or concentrated tubular fluid. The loop of Henle, in particular, the thick ascending limb, is the major site where solute and water are separated. Consequently, excretion of both dilute and concentrated urine requires normal function of the loop of Henle.
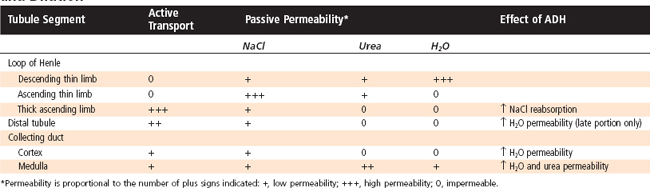
Figure 34-4 summarizes the essential features of the mechanisms whereby the kidneys excrete either dilute or concentrated urine. Table 34-3 summarizes the transport and passive permeability properties of the nephron segments involved in these processes.
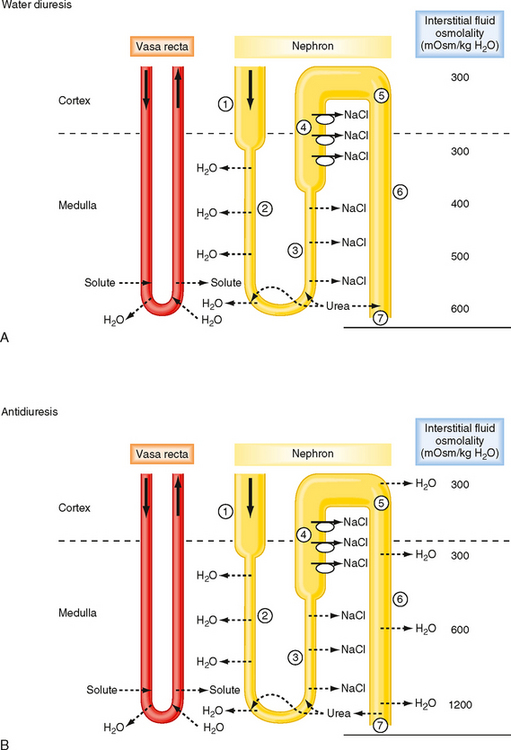
Table 34-3 Transport and Permeability Properties of Nephron Segments Involved in Urine Concentration and Dilution
First, how the kidneys excrete dilute urine (water diuresis) when ADH levels are low or zero is considered. The following numbers refer to those encircled in Figure 34-4, A:
Next, how the kidneys excrete concentrated urine (antidiuresis) when plasma osmolality and plasma ADH levels are high is considered. The following numbers refer to those encircled in Figure 34-4, B:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


