The constitutional hematopoietic disorders are a heterogenous group with regard to clinical presentation and pathology. Genetically, these disorders may be inherited or arise de novo. Most are quite rare. Interestingly, some of these disorders also occur as acquired syndromes, in the course of myelodysplastic syndromes and acute leukemia. Constitutional storage disorders are discussed in Chapter 8.
In most of these disorders, the underlying genetic defect has been found. Nevertheless, it is important to recognize that in many, genotype does not predict phenotype. Thus, there is much more to learn about the pathobiology of these syndromes.
Clinically, many patients have associated somatic malformations or other nonhematopoietic signs and symptoms. Some are characteristically diagnosed in neonatal life, others at any time from infancy through old age, and a few are recognized only in retrospect after a malignancy has arisen.
Pathologically, one or more hematopoietic cell lines may be affected. The following organization is based on the predominant lineage(s) affected. However, it should be kept in mind that this is not a strict grouping according to etiology or pathology, but simply a convenient way of categorizing constitutional hematopoietic disorders, and that considerable overlap exists among these categories. It should also be noted that some cases met in practice may not fit into any currently recognizable category.
CONSTITUTIONAL BONE MARROW FAILURE SYNDROMES
The inherited bone marrow failure syndromes are genetic disorders characterized by defective hematopoiesis (Table 4.1). One or more hematopoietic cell lines may be affected. A common feature in many cases is defective ribosomal synthesis, as found in cartilage-hair hypoplasia, Diamond-Blackfan anemia, dyskeratosis congenita, and Shwachman-Diamond syndrome. The centrality of ribosomal synthesis in cell development and maintenance helps account for the wide variety of clinical presentations and outcomes in these patients.
These syndromes tend to present in infancy or early childhood with anemia and other signs of bone marrow failure of variable severity and progression. They are often accompanied by skeletal and other malformations. The clinical course is marked by a tendency to the eventual development of myelodysplastic syndromes and malignancy including both hematopoietic and solid tumors.
Cartilage-Hair Hypoplasia
Cartilage-hair hypoplasia (CHH) is transmitted as an autosomal recessive trait (1, 2, 3, 4, 5, 6, 7). CHH is caused by a variety of different mutations of RMRP (for RNA component of mitochondrial RNA processing endoribonuclease), located on chromosome 9p21-p12 (1, 2, 3, 4, 5, 6, 7). One or both RMRP alleles may be mutated. The gene product is a subunit of the RNase MRP enzyme complex, a structure involved in many RNA processing events.
TABLE 4.1 Constitutional Bone Marrow Failure Syndromes
Cartilage-hair hypoplasia
Diamond-Blackfan anemia
Dyskeratosis congenital
Fanconi anemia
Griscelli syndrome
Shwachman-Diamond syndrome
Clinically, CHH is characterized by metaphyseal chrondrodysplasia, short stature, fine sparse hair, combined immunodeficiency, and hematopoietic abnormalities. Anemia is found in more than 80% of patients. In most patients, anemia is mild and transient; in less than 10%, it is severe and persistent. Chronic neutropenia may be present. Some cases show thrombocytosis. Little is known about the bone marrow pathology.
Diamond-Blackfan Anemia
Diamond-Blackfan anemia (DBA) is a genetically heterogeneous disorder usually transmitted as an autosomal dominant trait (8, 9, 10, 11, 12, 13). Twenty-five percent of patients bear a mutation of RPS19 (for ribosomal protein RPS19), located on chromosome 19q13.2. RPS19 is an essential component of the ubiquitous 40S small ribosomal subunit. Abnormalities in this protein affect ribosome biogenesis and nucleolar organization. In the remainder of cases, the genetic defect is still unknown.
Patients typically present during infancy with anemia. Congenital malformations, predominantly of the head and upper limbs, occur in 33% of cases. Hydrops fetalis has been reported.
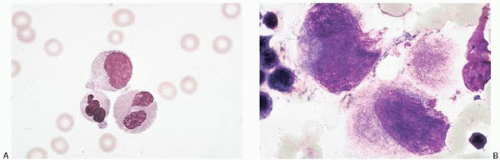
Figure 4.1 (A) Diamond-Blackfan anemia, peripheral blood. Neutrophils show dysplastic changes. (B) Diamond-Blackfan anemia, bone marrow aspirate. Dysplastic megakaryocytes are present.
Hematologic findings are highly variable, ranging from macrocytosis without anemia to overt congenital hypoplastic anemia (Fig. 4.1). Other peripheral blood findings include neutropenia, thrombocytosis, and, less often, thrombocytopenia, which may be present at diagnosis or develop during the course of the disease. The bone marrow shows erythroid hypoplasia, progressing to aplastic anemia. Other marrow findings include trilineal dysplasia, megakaryocytic hyperplasia, monocytosis, erythrophagocytosis, and fibrosis. Myelodysplastic syndrome and acute leukemia are late complications.
The differential diagnosis includes acquired erythroid hypoplasia in childhood in combination with an underlying hemolytic disease as seen in transient erythroblastopenia of childhood and in chronic parvovirus B19 infection (14,15).
Dyskeratosis Congenita
Dyskeratosis congenita (DC) is a genetically heterogeneous disorder transmitted predominantly in autosomal dominant and X-linked forms. In some cases the genetic mechanisms are unknown (16, 17, 18, 19, 20, 21).
Autosomal dominant DC is characterized by mutations in TERC (for telomerase RNA component), located on chromosome 3q26. The encoded protein is the reverse transcriptase component of telomerase. Both types of DC are characterized by genetic haploinsufficiency and premature and accelerated telemore shortening. Thus, the most affected tissues are those with the highest proliferative rate: skin, mucous membranes, and bone marrow. Pathology evolves over time with the accumulation of cell cycles.
X-linked DC is caused by a variety of mutations in DKC1 (for dyskeratosis congenita 1, dyskerin), located on chromosome Xq28. The gene product is an essential component of mRNA and telomere function, and its mutation is associated with premature telomere shortening.
The age of onset, type, and severity of DC lesions are highly variable. Males with X-linked DC usually present at age 10 years or later with skin hyperpigmentation, nail dystrophy, and mucous membrane leukoplakia, followed by bone marrow failure. This sequence is sometimes reversed, with presentation of DC in early childhood as bone marrow failure without ectodermal abnormalities. Other findings include neurologic dysfunction, restrictive pulmonary disease, and severe immunodeficiency. Females heterozygous for the DKC1 mutation show some clinical features of DC and marked skewing of X-chromosome inactivation, selecting for the normal DKC1 allele. X-linked Hoyeraal-Hreidarsson syndrome is the severe infantile variant of X-linked DC.
The peripheral blood shows cytopenias, especially anemia, neutropenia, and lymphopenia. The bone marrow shows progressive trilineal hypoplasia, culminating in aplastic anemia in more than 90% of cases by the age of 20. Dyserythropoiesis and dysmegakaryopoiesis have been reported. Myelodysplastic syndrome and acute leukemia are late complications. Death results from hematopoietic failure, pulmonary disease, and malignancy.
The differential diagnosis includes childhood myelodysplastic syndromes and aplastic anemia, which may be associated with acquired TERC mutations (22,23).
Fanconi Anemia
Fanconi anemia (FA) is a genetically heterogeneous group of autosomal recessive disorders, linked by chromosomal instability (24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39). Many different mutations have been described, affecting highly conserved genes that encode functionally related proteins. In general, these proteins are critical for DNA damage response, cell cycle regulation, and cell survival. The numerous genetic variants of FA are recognized as complementation groups; in many of these groups the affected gene has been identified. Interestingly, BRCA1 and BRCA2, more widely appreciated as genes conferring increased risk of breast cancer, are involved in some cases of FA. Patients with FA and biallelic mutations of BRCA2 have particularly early onset of anemia and acute leukemia.
FA is diagnosed from birth through adulthood, sometimes not until the development of acute leukemia. Patients present with cytopenias, hyperpigmentation, and a wide spectrum of congenital anomalies primarily involving the genitourinary and gastrointestinal tracts, limbs, and skeleton. FA patients are particularly susceptible to the development of hematopoietic disease (aplasia, myelodysplastic syndrome, acute leukemia) following chemotherapeutic agents used in the treatment of malignancies. Some cases of FA are not recognized until after the diagnosis of a malignancy has been made and therapy begun.
Chromosome instability may be detected in the laboratory by culturing patient lymphocytes with a DNA crosslinking agent (e.g., mitomycin C). Most, but not necessarily all, cultured metaphase chromosomes show excessive and abnormal breaks.
The peripheral blood shows cytopenias in 70% of patients by age 7 and in nearly 100% by age 40 (Fig. 4.2). Red blood cells show aniso- and poikilocytosis and may include teardrop cells and basophilic stippling. The bone marrow shows trilineal hypoplasia, progressing to aplastic anemia. Other findings include dyserythropoiesis and reactive mastocytosis.
Major complications include aplastic anemia, myelodysplastic syndromes, acute leukemia, and solid tumors. Hematopoietic clones arise frequently, showing expansion, evolution, regression, and even reversion to a normal genotype. By age 40, clonal hematopoiesis is found in nearly 100% of patients and recognizable myelodysplastic syndrome or acute leukemia in 50%.
Griscelli Syndrome
Griscelli syndrome (GS) is transmitted as an autosomal recessive disorder, usually caused by mutations in RAB27A, located on chromosome 15q15-q21 (40, 41, 42, 43, 44, 45). The gene product is a member of the RAS oncogene family, which is apparently required for normal melanosome formation and immunity.
Patients present with pigment dilution, which produces silvery-gray hair and hypopigmented skin. Other significant findings include pancytopenia, immunodeficiency, and failure to thrive.
The bone marrow has been reported to show hypercellularity. Myelodysplastic syndrome has been reported.
Complications may be lethal. GS is one of the causes of familial hemophagocytic lymphohistiocytosis, and patients typically progress to an accelerated phase of fatal hemophagocytic syndrome.
The differential diagnosis includes Chediak-Higashi disease, which shows abnormalities of pigment-containing cells and immunity; and Hermansky-Pudlak syndrome, which shows albinism and a bleeding disorder.
Shwachman-Diamond Syndrome
Shwachman-Diamond syndrome (SDS) is an autosomal recessive disorder, caused in most cases by mutation of one or both alleles of SBDS (for Shwachman-Bodian-Diamond syndrome), a highly conserved gene located on chromosome 7q11.21 (46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58). SBDS is usually found in the nucleus; its function is not known. As in many hematopoietic disorders, genotype is not necessarily predictive of phenotype.
Patients usually present shortly after birth with recurrent infection, pancreatic exocrine insufficiency, metaphyseal chondrodysplasia and other skeletal abnormalities, and growth retardation. Rarely, SDS presents as congenital aplastic anemia.
Laboratory studies show multiple functional abnormalities of neutrophils, erythroid precursors, and lymphocytes and increased hemoglobin F, findings consistent with a a stem cell disorder.
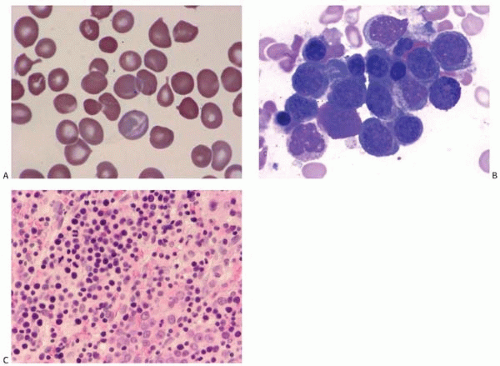
Figure 4.2 (A) Fanconi anemia, peripheral blood. Anisoand poikilocytosis, teardrop cells, and polychromasia are present. (B) Fanconi anemia, bone marrow aspirate. Erythroid hyperplasia with subtle dyserythropoiesis is seen in this patient with Fanconi anemia developing refractory anemia with ringed sideroblasts. (C) Fanconi anemia, bone marrow biopsy. Erythroid hyperplasia and numerous early erythroid precursors are present.
The peripheral blood shows neutropenia in all patients, anemia in 66%, and thrombocytopenia in 25%. Neutropenia may be constant or intermittent. Persistent neutropenia has been reported in 43% of patients. The bone marrow shows decreased CD34-positive cells, increased CD5-positive precursor B cells, and prominent apoptosis.
Complications include sepsis and clonal hematopoietic disorders, the latter seen in one third of patients. Isochromosome 7 is a common acquired clonal abnormality of uncertain clinical significance. Patients have an increased risk of myelodysplastic syndromes and acute myeloid leukemia. In some cases, clonal cell populations have apparently regressed. The differential diagnosis includes congenital cyclic neutropenia, Kostmann disease, and severe congenital neutropenia.
CONSTITUTIONAL ERYTHROCYTE DISORDERS
The constitutional (familial, hereditary) erythrocyte disorders may exhibit excess erythrocyte production, causing erythrocytosis; or decreased erythrocyte production, causing functional or true anemia (Table 4.2). The majority of erythrocyte disorders are characterized by decreased, ineffective, and’or dysplastic erythropoiesis. This group of disorders may be diagnosed at any time in life, from infancy to old age.
Constitutional Erythrocytosis
Constitutional erythrocytosis or polycythemia is a genetically heterogeneous group of disorders characterized by inappropriate erythrocytosis (59, 60, 61). These disorders may be found at birth or at any other time of life. They may be traced to abnormal erythropoietin (EPO) sensitivity of erythroid precursors, abnormal EPO regulation, abnormalities intrinsic to mature red blood cells, and other etiologies. This discussion is limited to abnormalities of EPO sensitivity and regulation.
TABLE 4.2 Constitutional Erythrocyte Disorders
Constitutional erythrocytosis
Congenital dyserythropoietic anemia
Constitutional megaloblastic anemia
Constitutional disorders of iron metabolism
Congenital sideroblastic anemia
Erythrocyte membrane abnormalities
Erythrocyte enzymopathies
Abnormal hemoglobins
Unbalanced globin chain production (thalassemia)
Chuvash polycythemia is one of the best-studied types of constitutional erythrocytosis (62, 63, 64, 65, 66, 67, 68, 69, 70). It was originally described as an endemic disorder in the Chuvash region of Russia, and has since been found as an endemic disorder in the island of Ischia and as a sporadic disorder in other locations and ethnic groups. It occurs in patients with various heterozygous, compound heterozygous, and homozygous mutations of VHL (for von Hippel-Lindau tumor suppressor), located on chromosome 3p26-p25. VHL is a component of an enzymatic complex that targets hydroxylated hypoxia-inducible factor (HIF) for degradation. HIF is, in turn, an important component of the oxygen-sensing pathway based in the kidney. Abnormalities of VHL interfere with normal HIF degradation. This leads to inappropriately increased HIF, followed by increased EPO levels and finally erythrocytosis. Patients with VHL mutations may also suffer from an increased incidence of thrombosis, vascular abnormalities, and early death.
A second form of constitutional erythrocytosis, accounting for approximately 15% of cases, is transmitted as an autosomal dominant disorder (71, 72, 73). It is characterized by mutations of EPOR, located on chromosome 19p13.3-p13.2, which encodes the EPO receptor. EPOR mutations result in truncated forms of the EPO receptor, which in turn causes EPO hypersensitivity in erythroid progenitors. EPO hypersensitivity results in erythrocytosis.
A third form of constitutional erythrocytosis is caused by mutation of EGLN1 (for egl nine homolog 1), located on chromosome 1q42.1. The gene product is an enzyme that hydroxylates HIF (74,75). As described above, HIF is an important component of the oxygen-sensing pathway in the kidney. Abnormal forms of EGLN1 interfere with normal HIF degradation, and thus produce inappropriately elevated EPO levels and erythrocytosis.
Figure 4.3 (A) Congenital dyserythropoietic anemia, peripheral blood. A binucleated red blood cell is present. (B) Congenital dyserythropoietic anemia, bone marrow biopsy. Erythroid hyperplasia is present.
Lastly, some cases of constitutional erythrocytosis are apparently caused by an as-yet-undefined genetic abnormality, located on chromosome 7q22 (76).
Patients with constitutional erythrocytosis are affected at birth, but the diagnosis may be made at any age when unexpected erythrocytosis is discovered. The genotype does not predict phenotype. The red blood cell count, red blood cell mass, and hematocrit are increased. The serum EPO level is variable and the arterial oxygen saturation is normal. The white blood cell and platelet counts are normal. The peripheral blood shows no morphologic abnormalities. The bone marrow shows erythroid hyperplasia.
The differential diagnosis includes high oxygen-affinity hemoglobinopathy and 2,3-bisphosphoglycerate deficiency, discussed below with abnormal hemoglobins and enzymopathies, respectively; neonatal erythrocytosis associated with trisomy 21 (Down syndrome) (77); other cases of neonatal erythrocytosis (78); a wide spectrum of acquired disorders and idiopathic erythrocytosis, discussed in Chapter 4; and myeloproliferative disorders.
Congenital Dyserythropoietic Anemia
Congenital dyserythropoietic anemia (CDA) comprises several rare disorders, usually presenting in infancy, characterized by abnormal and ineffective erythropoiesis (Fig. 4.3) (79, 80, 81). Most, but not all, fall into one of the three groups termed CDA I, II, and III. In addition to these entities are Majeed syndrome and isolated, unclassified cases of CDA.
CDA type I is transmitted as an autosomal recessive disease. It is caused by various mutations of CDAN1, located on chromosome 15q15 (82, 83, 84, 85, 86, 87, 88). The gene product is a protein that shows some similarities to microtubule-associated proteins and may be important in the formation of the nuclear envelope and the skeleton. The diagnosis is usually made at birth and occasionally in adulthood. Newborns and older subjects may present with severe, chronic anemia and reticulocytopenia; transient thrombocytopenia; jaundice, hyperbilirubinemia, and gallstones; splenomegaly; persistent pulmonary hypertension; and bony abnormalities. A thalassemia-like abnormality has also been reported. Hemochromatosis eventually develops, exacerbated by transfusion and’or coinheritance of Gilbert syndrome. The peripheral blood shows macrocytic anemia with marked anisopoikilocytosis, polychromasia, and basophilic stippling. The bone marrow demonstrates marked erythroid hyperplasia. Erythroid precursors exhibit megaloblastic change, binucleation, internuclear bridging, and uneven heterochromatin condensation, ultrastructurally appearing spongy or Swiss cheese-like. Ringed sideroblasts may be present but are not characteristic.
CDA type II (formerly known as HEMPAS, hereditary erythroblastic multinuclearity with a positive acidified serum test) is transmitted as an autosomal recessive disease and is the most frequent of the CDA syndromes (89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99). It is usually caused by a mutation of CDAN2, located on chromosome 20q11.2-q11.2, which leads to defective glycosylation of red blood cell membrane proteins. One case has been described in which CDAN2 was not mutated but the glycosylation defect was nevertheless present. The diagnosis is usually made at birth or during childhood. Patients present with anemia and eventually develop pigment cholelithiasis, splenomegaly, and hemochromatosis with its complications. Iron overload may be exacerbated by transfusion and coinheritance of other hematopoietic diseases, hereditary hemochromatosis, and Gilbert syndrome. Laboratory testing typically, but not always, shows a positive acidified serum test. The peripheral blood shows normocytic to macrocytic, hemolytic anemia with microspherocytes. The bone marrow shows erythroid hyperplasia with bi- and multinucleated erythroid precursors. Aplastic anemia has been reported in CDA II patients with parvovirus B19 infection. The differential diagnosis includes hereditary spherocytosis, which differs in showing reticulocytosis rather than reticulocytopenia.
CDA type III is transmitted as an autosomal dominant disease (100, 101, 102, 103, 104). It is caused by a mutation of CDAN3, located on chromosome 15q21-q25. The function of this gene is not known. The diagnosis may be made at any age. Patients may present with heart failure due to anemia and’or congenital cardiac abnormalities, hepatosplenomegaly, and retinal abnormalities. The peripheral blood shows mild to moderate macrocytic hemolytic anemia. The bone marrow shows erythroid hyperplasia with giant, multinucleated erythroid precursors. An increased incidence of monoclonal gammopathy and multiple myeloma has been reported in these patients.
A fourth type of CDA is a syndrome comprised of congenital dyserythropoietic anemia, chronic recurrent multifocal osteomyelitis, and neutrophilic dermatosis (Majeed syndrome) (105, 106, 107). It is transmitted as an autosomal recessive disorder and is caused by a mutation in LPIN2 (for lipin 2), located on chromosome 18p11.31. The encoded protein apparently plays a role in fat and triglyceride metabolism. Neonates present with anemia and hepatosplenomegaly, followed by recurrent bouts of fever, bone pain, osteomyelitis, and inflammatory skin lesions consistent with Sweet syndrome. The inflammatory manifestations in bone and skin are considered to be autoimmune in etiology. The peripheral blood shows hypochromia and microcytosis. The bone marrow shows significant dyserythropoiesis.
Other isolated and unclassified cases of CDA have also been reported. These include CDA with intraerythrocytic precipitations of a nonglobin protein (108); CDA with ringed sideroblasts, microcephaly, and intrauterine growth retardation (109); a syndrome similar to CDA II with hydrops fetalis (110); and other unusual, unclassified cases (111,112). Other reports of sporadic, unclassified CDA may be found in the literature, often in association with other somatic abnormalities.
The differential diagnosis of CDA includes other congenital dyserythropoietic disorders, such as GATA1-related dyserythropoietic anemia with macrothrombocytopenia (discussed below), and acquired dyserythropoiesis. The latter may be nonclonal, as in copper deficiency, parvovirus B19 infection, and other disorders (discussed in Chapter 16); or clonal, as found in acute myeloid leukemia, myelodysplastic syndromes, and myeloproliferative disorders.
Constitutional Megaloblastic Anemia
Constitutional megaloblastic anemia (CMegA) encompasses a heterogeneous group of autosomal recessive conditions affecting cobalamin (vitamin B12) metabolism (113).
One form of CMegA, selective vitamin B12 malaborption with proteinuria (Imerlund-Gräsbeck syndrome), is caused by mutations of CUBN, located on chromosome 10p12.31, and AMN, located on chromosome 14q32.3 (114,115). These genes encode cubilin (intrinsic factorcobalamin receptor) and amnionless protein, respectively, which act together in the ileal epithelium to absorb the cobalamin-intrinsic factor complex, and in the renal tubular epithelium to facilitate protein resorption.
A second form of CMegA is caused by a mutation in GIF, located on chromosome 11q13, which results in congenital absence of gastric intrinsic factor (116,117).
Other less common forms of CMegA include thiamine-responsive megaloblastic anemia syndrome with diabetes and deafness (118), defective cobalamin synthesis associated with hemolytic-uremic syndrome (119), hereditary transcobalamin II deficiency (120), and other rare inborn errors of metabolism (121,122), including mitochrondrial cytopathy (123).
The peripheral blood shows profound pancytopenia with macro-ovalocytes and hypersegmented neutrophils. The bone marrow shows marked megaloblastic hematopoiesis.
The differential diagnosis includes neonatal megaloblastic anemia acquired through maternal nutritional cobalamin deficiency (124), and megaloblastic anemia acquired later in life, discussed in Chapter 6.
Constitutional Disorders of Iron Metabolism
Anemia due to genetic defects in iron metabolism are rare diseases, transmitted as autosomal recessive disorders.
Congenital atransferrinemia, the more common of these disorders, is caused by a mutation in TF (for transferrin), located on chromosome 3q22.1 (125). TF binds iron and unites with the TF receptor to form the TF-TF receptor complex, which is then taken into duodenal epithelial cells via endosomes. TF mutation prevents the initial step in this pathway.
A second congenital disorder is caused by mutation of SLC11A2, located on chromosome 12q13, which encodes the solute carrier family 11, member 2 (126). This protein is a proton-coupled divalent metal ion transporter. Once the TF-TF receptor complex is taken into epithelial cells, SLC11A2 mediates the transfer of divalent iron from the endosome to the cytosol. SLC11A2 mutation prevents this step from occurring.
The peripheral blood shows severe hypochromic, microcytic anemia. The bone marrow shows erythroid hyperplasia with increased iron stores, a finding which distinguishes this disorder from iron-deficiency anemia. Other organs show marked excessive iron deposition.
The differential diagnosis of microcytic anemia with iron overload includes aceruloplasminemia (127,128), among other constitutional anemias (129, 130, 131), and acquired anemia of chronic inflammation or disease, discussed in Chapter 5.
Congenital Sideroblastic Anemia
Congenital sideroblastic anemia is a broad term encompassing a heterogeneous and complex group of anemias (Table 4.3) (132,133). Modes of transmission include mitochondrial, X-linked, autosomal recessive, an unusual mixed mitochondrial and autosomal recessive form, and other undefined pathways. In general, sideroblastic anemia arises when mitochondria malfunction, disturbing the oxidative pathway essential for cellular function. Their end result is a deficiency of intracellular adenosine triphosphate (the main cellular energy source) and uncontrolled intracellular release of damaging free oxygen radicals. Mitochondrial dysfunction is apparent as intramitochondrial iron deposition within the erythroid precursor, the hallmark “ringed sideroblast.”
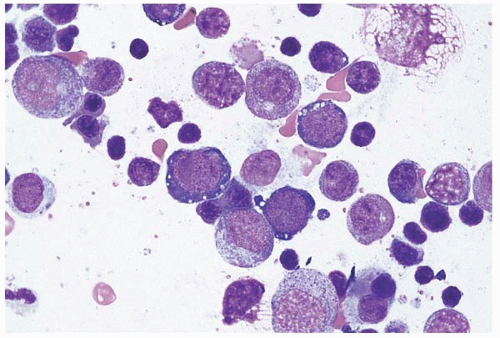
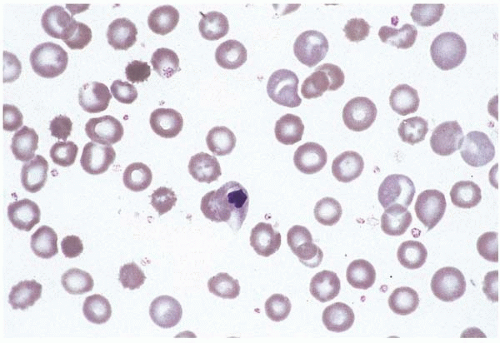
Figure 4.4 Pearson syndrome, bone marrow aspirate. The erythroid precursors are vacuolated.
Mitochondrial cytopathy comprises a genetically heterogeneous group of disorders in which mitochondrial DNA shows deletions, duplications, and other mutations, of either sporadic or inherited origin (Fig. 4.4) (134, 135, 136, 137, 138, 139, 140, 141). The clinical course is highly variable but usually progressive and ultimately fatal. Genotype, as in several other inherited hematopoietic diseases, does not necessarily predict phenotype (142). Mitochondrial cytopathy presents in the neonate as anemia and, in most cases, pancreatic insufficiency (Pearson marrow-pancreas syndrome). The peripheral blood shows severe macrocytic anemia, sometimes accompanied by neutropenia and’or thrombocytopenia. The bone marrow shows erythroid and myeloid precursors with distinctive vacuolation and ultrastructurally abnormal mitochondria. Ringed sideroblasts and increased iron stores are usually present. A rare case of megaloblastic anemia has been described (143). Survivors and those presenting later in life develop progressive hematopoietic, neuroendocrine, and other systemic organ failure (Kearns-Sayre syndrome).
X-linked sideroblastic anemia (XLSA), the most common inherited SA, is caused by a mutation of ALAS2 (for aminolevulinate, delta-, synthase 2), located on chromosome Xp11.21 (144, 145, 146, 147, 148, 149, 150, 151, 152, 153). The gene product is located within mitochondria, where it catalyzes the rate-limiting step in heme synthesis. Thus, XLSA has features of both an X-linked disorder and a mitochondrial cytopathy, complicated by variable penetrance. Anemia may be diagnosed at any age and in either gender. Disease in females is possible through mutations in both X chromosomes and, in older women, age-related skewing of X-chromosome inactivation in erythroid precursors. Hemochromatosis may be present because of transfusional iron overload and’or coinheritance of other hematopoietic disorders. The peripheral blood shows mild to severe microcytic hypochromic anemia. Occasionally, anemia is absent. The bone marrow shows erythroid hyperplasia with numerous ringed sideroblasts. Disease progression may occur, resulting in severe iron overload and its complications. Anemia is ameliorated by pyridoxine therapy in about 90% of cases.
XLSA with ataxia (XLSA’A) is caused by a mutation of ABCB7 (for ATP-binding cassette, sub-family B, member 7), located on chromosome Xq12-q13 (154, 155, 156, 157, 158). The gene product is believed to transport iron from the mitochondrion to the cytoplasm. Thus, XLSA’A has characteristics of both an X-linked disorder and a mitochondrial cytopathy. Male infants present with anemia and spinocerebellar ataxia. The peripheral blood shows mild hypochromic, microcytic anemia with Pappenheimer bodies. The bone marrow shows erythroid hyperplasia with ringed sideroblasts. Carrier females are not anemic but the peripheral blood may show red blood cell dimorphism, hypochromia, basophilic stippling, and’or ringed sideroblasts.
Mitochondrial myopathy and sideroblastic anemia (MLASA) is a recently described entity with features of both a mitochondrial cytopathy and an autosomal recessive condition (158, 159, 160, 161, 162). MLASA is caused by a mutation of PUS1 (for pseudouridylate synthase 1), located on chromosome 12q24.33. The encoded protein is present in both the nucleus and mitochondria. It converts uridine into pseudouridine in both mitochondria and cytosol, thus the clinical findings of both a mitochondrial myopathy and an autosomal recessive disorder. Patients present with onset of sideroblastic anemia at adolescence, lactic acidemia, and muscle weakness.
Other rare, unclassified cases of congenital sideroblastic anemia with dyserythropoiesis have been reported (163,164).
The differential diagnosis of congenital sideroblastic anemia includes acquired nonclonal sideroblastic anemia, as seen in copper deficiency (165,166); some forms of drug therapy (167); and acquired clonal sideroblastic anemia, a myelodysplastic syndrome. Of interest is the fact that a rare case of myelodysplastic syndrome has been shown to have mtDNA deletions identical to those reported in hereditary mitochondrial cytopathy (168).
Erythrocyte Membrane Abnormalities
The erythrocyte membrane abnormalities are a disparate group of disorders, characterized by abnormal erythrocyte morphology (Table 4.4) (169, 170, 171, 172). Some may confer resistance to malaria; however, this effect has been difficult to prove.
TABLE 4.4 Erythrocyte Membrane Abnormalities
Hereditary elliptocytosis
Hereditary pyropoikilocytosis
Hereditary spherocytosis
Dehydrated hereditary stomatocytosis
Overhydrated hereditary stomatocytosis
Southeast Asian ovalocytosis
Phytosterolemia (Mediterranean stomatocytosis and macrothrombocytopenia)
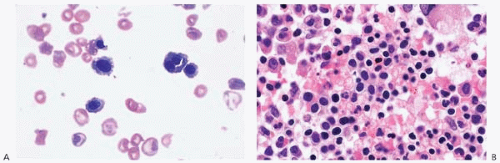
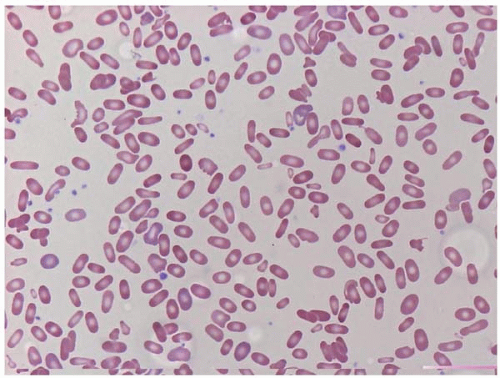
Figure 4.5 Hereditary elliptocytosis, peripheral blood. Numerous elliptocytes are present as well as microspherocytes and red blood cell fragments.
Hereditary elliptocytosis (HE) occurs as a sporadic disorder and as an inherited condition, with transmission as both an autosomal dominant and autosomal recessive disorder (Fig. 4.5) (173,174). The main populations affected are of African and Mediterranean origin. HE is caused by mutations and polymorphisms of genes encoding linkage proteins between the erythrocyte cytoskeleton and the cell membrane. These include SPTA1 (for spectrin, alpha, erythrocytic 1), which is located on chromosome 1q21 and encodes the spectrin a-chain; SPTB (for spectrin, beta, erythrocytic), which is located on chromosome 14q23-q24.2 and encodes the spectrin b-chain; and EPB41, which is located on chromosome 1p33-p32 and encodes erythrocyte membrane protein band 4.1. Mutations in these genes result in proteins which interfere with the formation of spectrin tetramers. The result is diminished membrane elasticity with consequent membrane destabilization, erythrocyte fragmentation and elongation (elliptocytosis), and increased osmotic fragility. Patients may be asymptomatic or present with hemolytic anemia, splenomegaly, and’or jaundice.
Laboratory studies show an increased red cell distribution width (RDW). The peripheral blood smear shows a spectrum of changes among patients, ranging from a simple predominance of elliptocytes to a wide variety of elliptocytes, spherocytes, and abnormally shaped RBC fragments. The latter appearance is nearly indistinguishable from findings in HPP and may suggest a diagnosis of HS. The differential diagnosis includes acquired elliptocytosis, a finding reported in myelodysplastic syndrome.
Hereditary pyropoikilocytosis (HPP) is a severe form of HE (175, 176, 177, 178, 179, 180). Studies of the original HPP kindred have shown a combination of a mutation and a polymorphism in SPTA1, which encodes the spectrin a-chain. The clinical findings range from asymptomatic elliptocytosis to severe hemolytic anemia and may change over time. An individual may have HPP at birth, then hemolytic HE, followed by asymptomatic HE.
Laboratory studies show extreme erythrocyte fragility and fragmentation upon incubation at 37 to 48°C; thus the designation “pyropoikilocytosis.” The peripheral blood smear shows elliptocytes, microspherocytes, polychromasia, nucleated red blood cells, and fragmented erythrocytes. Bizarre erythrocyte morphology and nucleated red blood cells are especially prominent in infants with HPP. After splenectomy, erythrocytes may appear spiculated rather than elliptical. The bone marrow shows marked ineffective erythroid hyperplasia, which may be accompanied by dyserythropoiesis.
The differential diagnosis includes neonatal hyperbilirubinemia associated with Rh incompatibility; erythrocyte abnormalities secondary to burns; microangiopathic hemolytic anemia; acquired pyropoikilocytosis seen in acute and chronic leukemia; and artifactual erythrocyte fragmentation induced by storage at high temperatures.
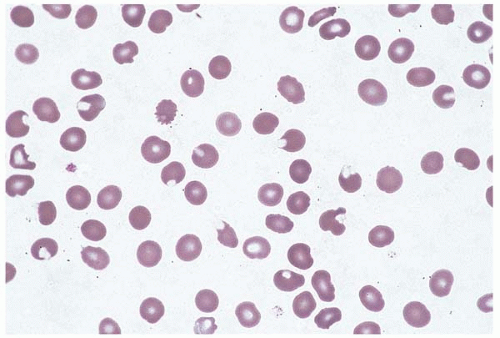
Hereditary spherocytosis (HS) occurs sporadically and as an inherited condition, with transmission as an autosomal dominant, autosomal recessive, and compound heterozygous disorder (Fig. 4.6) (181, 182, 183, 184, 185, 186, 187, 188, 189, 190, 191, 192, 193, 194, 195, 196). It has been described in Causasian and Japanese populations. HS is caused by abnormalities in the genes encoding the spectrin α-chain and spectrin β-chain described above in HE; as well as ANK1 (for ankyrin 1, erythrocytic), located on chromosome 8p11.1; SLC4A1 (for solute carrier family 4, anion exchanger, member 1), located on 17q21-q22 and encoding erythrocyte membrane protein band 3; and EPB42, located on chromosome 15q15-q21 and encoding erythrocyte membrane protein band 4.2. Absence or abnormalities in these proteins lead to microvesiculation or blistering of the membrane, microspherocytosis, and hemolysis, followed by splenic sequestration and destruction.
Figure 4.6 Hereditary spherocytosis, peripheral blood. Nearly all the red blood cells are microspherocytes or polychromatophilic cells.
Laboratory studies reveal mild to moderate hemolysis. Glycated hemoglobin (HbA1c) may be underestimated due to significant chronic hemolysis. The peripheral blood demonstrates microcytic, hyperchromic anemia, with polychromasia and microspherocytes proportional to the degree of anemia. The bone marrow shows marked erythroid hyperplasia. HS may be masked by concomitant folate, vitamin B12, or iron deficiency. It has been reported in combination with hemoglobin S, β-thalassemia, and hemochromatosis.
Complications include parvovirus B19 infection and subsequent aplastic crisis, extramedullary hematopoiesis, leg ulcers, and early cholelithiasis.
The differential diagnosis includes congenital dyserythropoietic anemia and immune-mediated hemolytic anemia.
Dehydrated hereditary stomatocytosis (DHS), or cryohydrocytosis, is an uncommon autosomal dominant disorder, reported primarily in northern European populations (197, 198, 199, 200, 201, 202, 203, 204, 205, 206, 207, 208, 209). It is characterized by a mutation in DHS, located on chromosome 16q23-q24. The mutation converts the encoded protein from an anion exchanger to a cation channel. The erythrocyte membrane is abnormally permeable to both sodium and potassium, an effect which is especially marked at or below room temperature. Laboratory abnormalities occurring after storage at or below room temperature include pseudohyperkalemia, increased osmotic fragility, pseudomacrocytosis, macrospherocytosis, and marked autohemolysis. The peripheral blood may show target cells, stomatocytes, and evidence of true (in vivo) or hemolytic anemia. Complications include polyhydramnios, self-limiting perinatal ascites, hepatitis, and hydrops in affected infants of mothers with DHS; parvovirus B19 infection with aplastic crisis; hepatic iron deposition (hemosiderosis); and thrombophilia following splenectomy. The differential diagnosis includes hereditary spherocytosis.
Overhydrated hereditary stomatocytosis (OHS) is a very rare disorder, apparently caused by a selective and partial deficiency of stomatin, an erythrocyte membrane protein (210, 211, 212, 213, 214). No genetic abnormality has yet been found in patients with OHS, and the pathophysiology of the disorder remains unclear. The peripheral blood shows evidence of hemolysis and macrocytosis. Affected infants may show a congenital syndrome of marked neurological abnormalities, cataracts, and hepatosplenomegaly. An increased risk of thrombosis following splenectomy has been reported.
Southeast Asian ovalocytosis (SAO) is transmitted as an autosomal dominant, autosomal recessive, and compound heterozygous condition (215, 216, 217, 218, 219, 220, 221, 222). It has been reported primarily from southeast Asia and Melanesia, but has also been found in an Amerindian Mexican population. The condition may have a protective effect against some forms of malaria but this has been difficult to prove. SAO is caused by a deletion, or less commonly a mutation, in SLC4A1 (for solute carrier family 4, anion exchanger, member 1), located on chromosome 17q21-q22. The encoded protein normally transports chloride and bicarbonate ions across the erythrocyte plasma membrane. Gene deletion or mutation results in loss of function and abnormal ionic permeability. Clinically, patients may present with hemolytic anemia and ovalocytes. Some patients also show distal renal tubular acidosis, based on the same loss of function. Newborns present with anemia, hemolysis, hyperbilirubinemia, reticulocytosis, increased red cell distribution width, increased mean corpusculor volume, and stomatocytic ovalocytes. SAO has been reported with other red blood cell abnormalities.
Phytosterolemia (Mediterranean stomatocytosis and macrothrombocytopenia; sitosterolemia) is a rare autosomal recessive condition characterized by abnormal plant sterol metabolism (223, 224, 225). Thus, it is not primarily a hematopoietic disease. It is caused by mutations in ABCG5 or ABG8 (named for ATP-binding cassettes 5 and 8, respectively), located on chromosome 2p21. These genes encode proteins expressed in intestinal epithelial cells as transporters of cholesterol and plant sterols, or phytosterols. Plant sterols compete with cholesterol for intestinal absorption and thereby lower serum cholesterol concentrations. Defects in the transporter molecules produce hyperabsorption and diminished biliary excretion of plant sterols.
Patients present with early atherosclerosis and its complications, xanthomas, hemolysis, and macrothrombocytopenia.
Laboratory studies show high levels of phytosterols in the blood (phytosterolemia), which may coexist with either normal or elevated blood cholesterol levels. The peripheral blood shows stomatocytic hemolysis and very large platelets, apparently due to the high blood level of phytosterol.
Erythrocyte Enzymopathies
Numerous erythrocyte enzyme deficiencies have been described (Table 4.5). The majority cause chronic nonspherocytic anemia. The two most common enzymopathies (G6PD and PK deficiencies) are discussed first, followed by the other hemolytic enzymopathies in alphabetical order. At the end are short discussions of an erythrocyte enzymopathy causing familial erythrocytosis rather than anemia (BPGM deficiency) and a nonerythrocytic enzymopathy that worsens the effects of the hereditary hemolytic anemias (UGT1A1 deficiency).
TABLE 4.5 Erythrocyte Enzymopathies
Glucose-6-phosphate dehydrogenase deficiency
Pyruvate kinase deficiency
Adenylate kinase deficiency
Glucose-6-phosphate isomerase deficiency
Glutathione synthetase deficiency
Hexokinase deficiency
Phosphofructokinase deficiency
Phosphoglycerate kinase deficiency
Pyrimidine 5′-nucleotidase type 1 deficiency
Triosephosphate isomerase 1 deficiency
2,3-Bisphosphoglycerate mutase deficiency
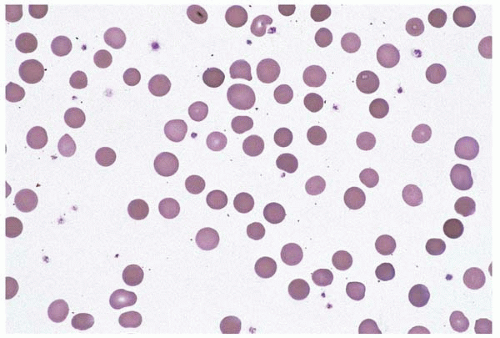
Figure 4.7 Glucose-6-phosphate dehydrogenase deficiency, peripheral blood. Numerous red blood cells with a peripheral crescentic defect (bite cells) are seen.
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is the most common enzyme deficiency worldwide, found in almost 20% of certain ethnic groups (Fig. 4.7) (226, 227, 228, 229, 230, 231, 232, 233, 234, 235, 236, 237, 238). It is caused by various mutations of G6PD, located on chromosome Xq28. G6PD catalyzes the first step of the pentose phosphate pathway, which converts glucose 6-phosphate to 6-phosphogluconate with production of NADPH2. Females may be affected because of the variable clinical impact of different mutations and the possibility of inheriting two different mutations (compound heterozygosity).
Clinical findings are variable and may be exacerbated by coinheritance of other genetic disorders and by parvovirus B19 infection. Most patients are asymptomatic. Acute hemolytic episodes may be triggered by fava bean ingestion (favism), oxidant drug exposure, vitamin C, butyl nitrite, fever, infection, metabolic acidosis, mothballs, henna, and numerous other oxidant stressors. Favism by proxy occurs in newborns and nursing infants through maternal ingestion of fava beans. Patients may also manifest chronic hemolytic anemia. Neonatal jaundice and hyperbilirubinemia are common.
The peripheral blood shows anemia and abnormal red cell morphology during a hemolytic episode. Hemighosts are erythrocytes with a spherical concentration of hemoglobin at one end and colorless, closely apposed membranes at the other. Bite cells are erythrocytes with one or more peripheral concavities. The bone marrow demonstrates erythroid hyperplasia during and after a hemolytic episode, except when infected with parvovirus B19, which may produce erythroid hypoplasia.
The differential diagnosis includes transient G6PD deficiency, as seen in typhoid fever (239), and acquired G6PD deficiency, described in a patient with acute myeloid leukemia (240).
Pyruvate kinase (PK) deficiency is a common autosomal recessive condition caused by a mutation of PKLR in combination with another mutation or PKLR polymorphism (Fig. 4.8) (241, 242, 243, 244, 245, 246). One case has been described as an apparently autosomal dominant condition. PKLR (for pyruvate kinase, liver and RBC) is located on chromosome 1q21. PK is essential in the glycolytic pathway of the erythrocyte. PK deficiency may protect against malarial infection.
Figure 4.8 Pyruvate kinase deficiency, peripheral blood. Red blood cells with variable morphology and a dysplastic erythroid precursor are seen.
Patients present with mild to severe chronic hemolytic anemia and eventually develop hemochromatosis and its complications, which may be exacerbated by transfusion and coinheritance of other hematopoietic disorders.
The peripheral blood shows evidence of nonspherocytic hemolytic anemia. The bone marrow shows marked but largely ineffective erythroid hyperplasia. Extramedullary hematopoiesis has been reported. PK deficiency may be masked by infection and leukocytosis, which cause transient increases in PK activity.
The differential diagnosis includes acquired 2,3-bisphosphoglycerate mutase (BPGM) deficiency, reported in a patient with acute myeloid leukemia (240).
Adenylate kinase (AK) deficiency is a rare autosomal recessive disorder caused by mutations of AK1 (for adenylate kinase 1), located on chromosome 9q34.1 (247, 248, 249). AK affects the interconversion of the adenine nucleotides ATP and ADP.
Laboratory studies may show increased intraerythrocytic 2,3-diphosphoglycerate (2,3-DPG) levels and decreased levels of other erythrocyte enzymes. The peripheral blood shows evidence of chronic hemolysis, including reticulocytosis. Concomitant G6PD deficiency has been reported. Splenectomy may ameliorate the hemolysis.
Patients present with severe, chronic, nonspherocytic hemolytic anemia and, in at least some cases, neurological delay.
The differential diagnosis includes acquired AK deficiency, reported in a patient with acute myeloid leukemia.
Glucose-6-phosphate isomerase (GPI) deficiency is an uncommon autosomal recessive disorder, the result of mutations in GPI (for glucose phosphate isomerase), located on chromosome 19q13.1 (250, 251, 252). GPI catalyzes the interconversion of fructose-6-phosphate and glucose-6-phosphate. The peripheral blood shows evidence of chronic nonspherocytic hemolytic anemia, of variable severity. GPI deficiency has been reported in combination with G6PD deficiency.
Glutathione synthetase (GS) deficiency is a rare autosomal recessive disorder caused by mutations and other abnormalities in GSS (for glutathione synthetase), located on chromosome 20q11.2 (253, 254, 255, 256). GS catalyzes a step in the gamma-glutamyl cycle important in the production of glutathione, a critical intracellular antioxidant. The effects of GS deficiency are most marked under conditions of oxidative stress. Patients may show a range of findings, from essentially asymptomatic compensated hemolytic anemia to severe hemolytic anemia, metabolic acidosis, neurological defects, and increased susceptibility to infection. The peripheral blood shows evidence of chronic nonspherocytic hemolysis, usually with anemia. Splenectomy has been reported to ameliorate anemia. Coinheritance of G6PD deficiency has been described.
Only gold members can continue reading. Log In or Register to continue