Name
Components
Color
Tissue uptake
Alizarin red
Red/light purple
Calcium
Congo red
Red
Amyloid
Elastin von Giesen
Fuchsin/picric acid
Black
Elastin fibers
Eosin
Red
Cytoplasm, collagen, muscle fibers
Fontana massa
Ammoniacal silver nitrate
Black
Melanin, argentaffin granules
Gold chloride
Pink-red
Nuclei
Pale pink
Cytoplasm
Hematoxylin
Blue
Nuclei
Luxol fast blue/cresyl violet
Luxol fast blue
Blue
Myelin
Cresyl violet
Turquoise
Erythrocytes
Purple
Nuclei and Nissl substance
Masson’s Trichrome
Iron-hematoxylin
Black
Nuclei
Biebrich scarlet-acid Fuchsin
Blue green
Collagen fibers
Fast Green FCF
Pink/red/brown
Cytoplasm
Scarlet
Erythrocytes, keratin, myelin
Light green
Mucin
Movat’s pentachrome
Iron-hematoxylin
Black
Nuclei, elastic fibers
Alcian blue
Yellow
Collagen, reticular fibers
Resorcin-Fuchsin
Blue
Ground substance, mucin
Woodstain Scarlet-Acid Fuchsin
Scarlet
Fibrin
Saffron
Red
Muscle
Oil Red O
Sudan Red 5B, C26H24N4O
Red
Lipids (only in frozen section)
Prussian blue
Blue
Iron/hemosiderin
Toluidine blue O
Tolonium chloride, C15H16N3S
Blue green
Nuclei
Red
Mast cell, cartilage
Von Giesen
Picric acid
Pink/deep red
Collagen
Acid Fuchsin
Yellow
Muscle, cytoplasm, erythrocytes, fibrin
Von Kossa
Silver nitrate
Black
Calcium salts
Nuclear fast red
Wright/Giemsa
Eosin
Red
Erythrocytes, eosinophils
Methylene blue
Blue
Basophils, lymphocytes
Violet/purple
Neutrophils, platelets
The specific three-dimensional configuration of molecules recognized by an individual antibody is called an antigenic determinant or epitope. Antibody binding to a cell-specific marker allows specific cell types to be recognized within a tissue. For example, CD68 (Cluster of Differentiation 68) is a 110-kD transmembrane glycoprotein that is highly expressed by human monocytes and tissue macrophages. CD68 antibody can be used to identify tissue macrophages by targeting CD68 antigen on macrophages. Numerous antibodies to cell-specific markers are commercially available, although the actual specificity of any particular marker may need to be confirmed by an individual user.
IHC can also be used to identify certain cellular processes. Replicating cells may be recognized by the use of antibodies to proliferation-specific markers, Ki67 or proliferating cell nuclear antigen (PCNA), or by injection of a synthetic nucleotide bromodeoxyuridine (BrdU) prior to sacrifice followed by staining using an anti-BrdU antibody [1]. Localization of antibody staining within the nucleus, cytoplasm, or cell membrane can be used to determine cell-signaling and protein activation states [2]. For example, the transcription factor protein complex nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) when inactive can be detected in the cytoplasm using an antibody to the p65 component of the NF-κB protein complex. After activation, NF-κB moves to the nucleus; therefore, nuclear immunostaining of p65 indicates that NF-κB has been activated. Likewise, antibodies may be raised against a phosphorylated form of protein such as phospho-extracellular signal-regulated kinase (pERK) or phosphorylated isoform of protein kinase C (PKC) epsilon, which do not recognize the non-phosphorylated form, allowing activation of the extracellular signal-regulated kinase (ERK) or PKC-epsilon pathway to be identified [3] (Fig. 10.1).
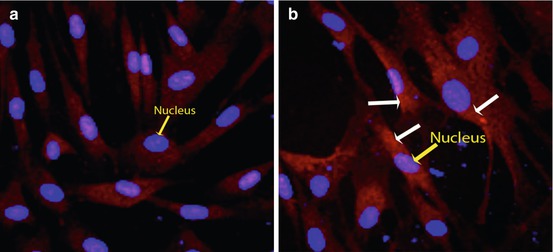
Fig. 10.1
Phosphoprotein labeling using fluorescence staining technique. (a) Background control without treatment shows no phosphorylated PKC epsilon. Blue nuclei are clearly demonstrated by the DAPI counterstain (yellow arrow). (b) Activation of PKC epsilon by phosphorylation (white arrows) is clearly identified following 30 min of 12-O-tetradecanoylphorbol-13-acetate treatment using a primary antibody specific to the phosphorylated form of PKC epsilon. A Texas red-conjugated secondary antibody was used for detection. The activated PKC epsilon can be localized to the cytoplasm, as the red signal does not overlap with the DAPI-counterstained blue nuclei (yellow arrow)
Apoptotic cells may be recognized by TUNEL staining (terminal deoxynucleotidyl transferase dUTP [2′-Deoxyuridine, 5′-Triphosophate] nick end labeling) or by antibody detection of cleaved caspase-3 or other apoptosis markers. The TUNEL assay relies on the presence of DNA fragments generated by apoptotic cells that can be identified by terminal deoxynucleotidyl transferase (TdT), an enzyme that will catalyze the addition of dUTPs that are secondarily labeled with a marker. The cleaved caspase assay, on the other hand, relies on caspase cleavage of protein substrates being a pivotal cascade that is unique to apoptotic cells [4, 5].
Oxidative stress can be detected either within the nucleus (anti-8-hydroxy-2′-deoxyguanosine or anti-thymidineglycol staining) [6], lipid membranes of cells (multiple oxidized lipid markers), or by protein oxidation (anti-tyrosine dimer, anti-nitrotyrosine, or anti-halogenated tyrosine) [7]. The downstream effects of oxidative stress may also be detected by IHC using antibodies for downstream signaling pathways such as the p65 component of NFκB [3]. The cytoskeleton and extracellular matrix can be studied using IHC. Antibodies to microtubules, actin, and intermediate filament components vimentin, keratin, and lamin are widely available, as are antibodies to collagen, elastin, and proteoglycan species [8]. Specific structural components such as focal adhesions can also be identified using antibodies to components of the focal adhesion complex such as vinculin or focal adhesion kinase [9].
Basic Principles
IHC was first described by Coons in 1941, using a fluorescent tagged antibody to identify pneumococci in frozen sections [10]. The technique was limited and did not enter widespread use until a number of different modifications increased its power and sensitivity, including enzyme linkage, peroxidase-antiperoxidase, avidin-biotin-peroxidase complex (ABC) or biotin-streptavidin, tyramide, and polymer-based labeling [11–17]. Antigen retrieval, developed in 1991 [18], significantly improved staining formalin-fixed, paraffin-embedded tissue sections, allowing widespread clinical use of IHC and more reliable immunostaining in specimens with better preserved architecture.
The antibodies used for clinical and research purposes belong to the immunoglobulin G (IgG) isotype and may be one of four subtypes, which becomes relevant when choosing the appropriate controls. Each antibody is comprised of two identical heavy chains and two identical light chains arranged in a Y-configuration. The ends of the Y comprise the variable region, which gives the antibody its specific binding properties, providing two antigen-binding sites per antibody. A smaller single antigen-binding domain (fragment antigen-binding or Fab fragment) can be generated by using papain to digest the antibody. The fragment crystallizable (Fc) portion of the antibody is species specific and contains domains for binding complement, the Fc receptor, Staph protein A, and inflammatory cells [19]. The different IgG subtypes vary in terms of these binding capabilities, with subtype 1 capable of binding all three binding domains, subtype 2 does not bind neutrophils, subtype 3 does not react with Staph A, and subtype 4 does not fix complement. Antibodies in one species can be raised to the Fc region of another species, allowing the amplification of signal. The first antigen-specific antibody is known as the primary antibody, as opposed to a species-specific secondary antibody [19].
When an animal is immunized with an antigen, it generates serum with multiple antibodies recognizing different epitopes at varying affinities, also known as polyclonal antibodies. Monoclonal antibodies are generated by fusing single antibody-producing B cells with non-antibody-producing myeloma cells, creating a hybrid cell line (hybridoma) which secretes a single antibody. The hybridomas are then screened for those that secrete an antibody that binds the desired antigen with appropriate specificity. Because monoclonal antibodies are generated by immortalized cell lines, there is usually good consistency from batch to batch. Furthermore, when purchasing an antibody, the specific clone is identified by the manufacturer, which can be compared to results published in the literature. Polyclonal antibodies are generally more sensitive but less specific than monoclonal antibodies and may have consistency issues from different batches. Antibodies may be raised against whole molecules or against the N-terminus, the C-terminus, specific amino acids, or phosphorylated states [19].
Techniques
There are multiple methodological approaches, but all involve three basic steps: sample preparation, sample labeling, and sample visualization. A sample protocol is shown in Fig. 10.2. Automated machines that can perform the vast majority of immunohistochemical protocols with the possible exception of antigen retrieval are available. However, the expense of the equipment generally means that they are only available in histological core and diagnostic pathology laboratories and are thus beyond the scope of this chapter.
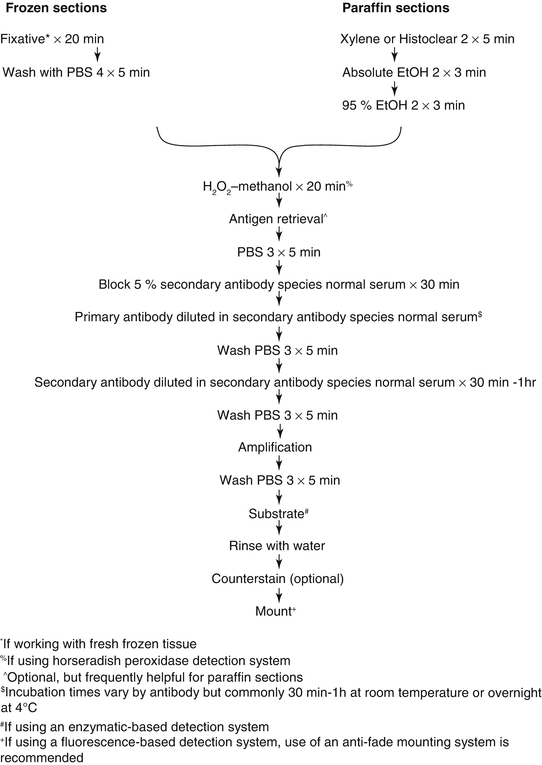
Fig. 10.2
Schematic of IHC protocol
Sample Preparation
Fixation
IHC builds on histochemistry by specific localization of target components within cells and tissue. This is only possible if the cells and tissues are appropriately processed and fixed such that they are recognizable under the microscope. Different fixation and tissue processing methods may affect antibody recognition such that strong staining is possible with one method, but not with another. In general, for research purposes, antibody staining is easier in frozen sections; however, tissue architecture is better in paraffin sections.
Because devitalized tissues undergo autolysis from release of lysosomal proteolytic enzymes, preservation of antigenicity requires either rapid fixation, which inactivates enzymes, or placing the tissue at sufficiently cold temperatures where enzymes are inactive. Fixation serves two additional purposes, to immobilize molecules within the cell and prevent diffusion and to increase tissue rigidity to improve sectioning. Antigen localization is optimized by more rapid fixation, either by improved delivery with perfusion fixation or by acceleration of the chemical process using heat or agitation [20]. Perfusion fixation has the additional advantage of improved vessel morphology. Small animals can be easily perfused at physiologic pressures through either the heart or the aorta using a sphygmomanometer or gravity-based perfusion fixation apparatus (Fig. 10.3).

Fig. 10.3
Perfusion fixation apparatus. Small animal pressure apparatus controlled with a pressure gauge. Alternatively the fixative and PBS flasks can be suspended overhead at a fixed level so as to provide a gravity-driven pressure equivalent to 100 mmHg. A peristaltic pump can also be used to exsanguinate the animal and circulate PBS to remove the blood components prior to perfusing with fixative. As the contents of the flasks are placed under pressure, an additional method of securing the rubber stoppers to prevent them from popping off the flask and potentially spraying toxic fixative is recommended. They are secured with wire in the pictured apparatus. The PBS is used to assist with exsanguination. (A) Flask for fixative. (B) Flask for PBS. (C) 3-way stopcock used to direct perfusion from either the fixative or the PBS flask. (D) 3-way stopcock used to allow administration of drugs or fluorescent dye. (E) Metal male luer which can be attached to an aortic catheter or blunt-tipped needle used to cannulate the left ventricle. (F) Pressure gauge. (G) Bulb to control pressure
As previously mentioned, freezing specimens generally leads to loss of tissue architecture due to the formation of ice crystals. Two strategies can be used for preservation of antigenicity while minimizing loss of architecture. One is to freeze the sample as rapidly as possible, by immersing it in isopentane cooled to its freezing point using liquid nitrogen, placing it on a metal block cooled by liquid nitrogen, or immersing it in some combination of dry ice and either acetone or ethanol. The other strategy is to use cryoprotection and dehydration. Sucrose is commonly used as a cryoprotectant [20]. For research purposes, specimens are generally embedded and frozen in optimal cutting temperature compound (OCT, mixture of polyvinyl alcohol and polyethylene glycol) to allow frozen sectioning. Cell membrane antigens and cytokines are frequently more successfully stained with frozen section rather than with paraffin-fixed tissue.
Chemical fixatives can be roughly divided between those that cause protein cross-linking and those that cause protein coagulation or precipitation. The most commonly used cross-linking fixative is formalin solution (37–40 % formaldehyde existing as low polymers and 60–63 % water). The low polymers of formaldehyde must be broken down to monomeric formaldehyde for active fixation by protein cross-linking through the aldehyde group, also known as a methylene bridge. This can be done by dilution with a buffer solution at physiological pH, generally to 4 or 10 %. Methanol (10 %) is frequently added to formalin by the manufacturer to slow down polymerization to paraformaldehyde. Paraformaldehyde, which is commonly used for research purposes, consists of higher polymers of formaldehyde and requires heating in buffered solution to 60 °C within a fume hood for solubilization [21]. Currently, both methanol-free formalin and aliquots of frozen formaldehyde prepared from paraformaldehyde are also commercially available. Although formaldehyde generally penetrates the tissue after 12–24 h, significant cross-linking may require up to 7 days to occur. As increased cross-linking tends to decrease antigenicity, over-fixation may render specimens unusable for immunohistochemical analysis.
Glutaraldehyde has two active aldehyde groups that allow more thorough cross-linking, making it ideal for electron microscopy, but less ideal for IHC as the free aldehyde groups must be removed or blocked to prevent nonspecific binding, and the thorough cross-linking frequently destroys antigenicity. The more thorough cross-linking also impedes paraffin penetration. From a practical standpoint, the more expensive “electron microscopy (EM) grade” glutaraldehyde that contains the monomer and low polymer forms required for adequate cell and tissue penetration rather than the “technical grade” glutaraldehyde that has larger polymers should be used for research purposes [21].
Coagulating fixatives include acetone, ethanol, methanol, trichloroacetic acid, and zinc-based fixatives. Acetone, ethanol, and methanol when used alone can cause significant shrinkage and hardening artifacts but do allow better preservation of DNA and RNA as well as preservation of antigenicity without the need for antigen retrieval. Carnoy’s fixative (60 % ethanol, 30 % chloroform, 10 % glacial acetic acid) or methyl Carnoy is a commonly used non-formalin fixative and may preserve carbohydrate and cytosolic membrane antigens more successfully than aldehyde-based fixatives [20].
Embedding and Sectioning
Whole mounts can be prepared for IHC without sectioning. They provide an immediate and three-dimensional view of stained antigens, which is useful for rapid surveillance. In general, the technique is limited to tissues that can be prepared as small blocks (<5 mm thick) such as embryos. Antibody penetration can be limited, however, resulting in patchy or false-negative staining.
Frozen tissue embedded in OCT may be cut immediately using a cryostat. Different tissues required cutting at different temperatures, which generally range between −20 and −30 °C. Slice thickness varies depending on the tissue and the skill of the operator but generally ranges between 5 and 10 μm. Thinner sections provide less possibility of cellular overlap and improved ultrastructural resolution. Certain applications, such as confocal microscopy, may call for considerably thicker sections. Cutting may introduce folds or wrinkles in the tissue, degrading its stainability and architecture. A newer method of tape transfer for frozen sections has been developed to improve tissue architecture. Essentially, tape is applied to the block prior to sectioning. After the section is cut, it is transferred tissue side down to a polymer-coated slide. The tissue is bonded to the slide using a flash of ultraviolet light, at which point the tape is removed, and the slides can then be processed as regular frozen sections for staining [22]. Tape transfer systems are commercially available for purchase, but the added expense may or may not be justifiable depending on the goals of the experiment. Slides should be maintained at −20 or −80 °C after cutting to maintain antigenicity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


