Fig. 7.1
Methods of protein purification. Various types of chromatography are shown in (a). Gel filtration chromatography (left panel) involves purification of proteins based on size. Small particles (red circles) become trapped in the small pores within resin beads, while larger particles (green circles) elute out quickly. Ion exchange chromatography (middle panel) involves using charged resin beads to purify protein. If the beads (black circles) are positively charged, the negatively charged proteins (white circles) will bind, while the positively charged proteins (red circles) will repel from the beads and filter through. Affinity chromatography (right panel) involves a lock and key-type mechanism of protein purification. The beads are coated with a binding partner or antibody to the target protein, which binds the desired protein to the column. All other proteins then elute out. The protein can then be dissociated from the column. (b) In SDS–PAGE, protein samples that have been exposed to SDS are loaded into the top wells of the gel. An electric field is then applied, and the proteins move toward the positive electrode. In the leftmost column is a protein standard which contains multiple proteins with known sizes. In this example, there are five remaining wells that contain unknown samples. The band corresponding to the protein of interest can be determined by comparing the bands to the ladder of protein standards. Since SDS-PAGE only separates proteins by size, each band may contain multiple proteins. To better separate an unknown sample, 2D gel electrophoresis may be used as shown in (c). First, proteins are separated by allowing movement through a pH gradient down a narrow strip of polyacrylamide gel (1). Proteins will be separated by their isoelectric point, or the pH at which their net charge is zero. The gel is then turned 90° and placed adjacent to a new piece of gel. The proteins are then exposed to SDS and an electric field is applied (2). Proteins will move toward the positive electrode as in SDS-PAGE. Thus, the final 2D gel resolves proteins based on isoelectric point along its width and size along its length
Gel filtration chromatography (Fig. 7.1a, left panel) simply involves separating proteins based on size. With this system, the column is composed of resin with certain sized pores. Small molecules fit inside the pores and are held up within the column, whereas larger proteins cannot fit in the pores and elute out quickly. As the solution slowly moves through the column, samples can be collected in separate tubes at various time points as they elute out the bottom. The different fractions collected each contain a relatively pure sample of proteins with similar sizes. If the size of the protein being studied is unknown, each fraction can be tested to determine which contains the desired protein by way of some of the methods discussed in the section on protein identification and quantification.
Hydrophobic interaction chromatography involves using resins within the column which have certain hydrophobic groups attached. As most proteins contain a variable number of hydrophobic regions, they will differentially adsorb to the resin, following the principal of “like dissolves like.” This process is promoted by using a salt-based solution that neutralizes most of the ionic charges on the protein and allows for increased interaction with the hydrophobic column. Other factors that promote adsorption of the protein to the column include temperature, pH, and concentration of the salt solution. The protein can then be eluted off the column by decreasing the ionic strength or adding other agents, such as denaturing solvents or detergents.
Ion exchange chromatography (Fig. 7.1a, middle panel) utilizes the ionic charge of the protein of study. Proteins that have an overall negative charge can be separated from a solution using a column containing a cationic resin and vice versa. The proteins are then eluted off by applying solutions with increasing ionic charges that act to displace the proteins adsorbed to the column.
Affinity chromatography (Fig. 7.1a, right panel) involves capturing the desired protein by attaching an enzyme, antibody, or protein-binding partner to the column. When a protein-containing solution is passed through the column, the desired protein will selectively bind to the column, while all other components will elute through. Examples of this method include “lock and key” interactions between an enzyme and its ligand, binding of glycoproteins to lectin, metal binding, and antibody-mediated binding (called immunoaffinity chromatography) [22, 23]. As described above, recombinant proteins may be constructed with a histidine tag, which has a high affinity for nickel and cobalt. Columns may be purchased which contain beads that are coated with either of these metals in order to purify His-tagged proteins. Other pre-made columns are coated with antibodies to specific fusion protein tags, such as GST. These binding interactions are quite strong but can be disrupted—thus eluting the protein from the column—by washing the column with concentrated salt solutions or solutions with a different pH.
High–performance liquid chromatography (HPLC) is a more modern method of chromatography that applies high pressures to the column to improve the resolution of the protein of study. In traditional chromatography, the flow of the protein-containing solution relies on the force of gravity. Thus, one of the main factors involved in the degree of purification is the spacing of the resin beads. If the beads are too tightly packed, the flow of the protein-containing solution is slower, and the degree of nonspecific binding increases. Alternatively, if the beads are not tight enough, the proteins will pass too quickly, and the yield of purified protein will be lower. In HPLC, the beads are packed exceedingly tight, making the flow of the protein solution close to zero. When high pressure is applied, the solution flows through the column in seconds as opposed to hours with traditional chromatography. Since the column beads are so compact, there is more surface area for protein binding, which allows for a much cleaner separation of the protein of study.
Gel Electrophoresis
Another preferred method of protein purification involves the application of electricity to a protein solution in a process entitled sodium dodecyl sulfate polyacrylamide gel electrophoresis (or SDS–PAGE) [3]. With this technique, proteins are first placed in SDS to unfold the proteins in solution. Most investigators also add a reducing agent, such as β-mercaptoethanol, to disrupt all disulfide bonds and fully denature the protein. SDS is a polar molecule with a hydrophobic chain on one end and a negatively charged sulfate group on the other end. As the amino acid side chains become exposed during protein unfolding, the hydrophobic ends of the SDS molecules bind to the hydrophobic portions of the denatured proteins, coating them with negative charges. The intrinsic charges on the proteins are thus masked by the large number of SDS molecules bound, and all proteins become net negatively charged.
The denatured proteins are then put through a highly cross-linked gel made from an inert substance, such as polyacrylamide, and an electrical field is applied (Fig. 7.1b). The negatively charged proteins then migrate through the gel toward the positive electrode. Larger proteins will be exposed to a larger electrical force, but due to the nature of the polyacrylamide matrix, they will have more drag and move slower through the gel. A solution of thousands of proteins can thus be separated into a series of discrete bands according to their molecular weight alone.
This process tends to be a more reliable way to separate proteins than those discussed previously, as it removes the variables of net ionic charge and protein shape to fractionate the proteins purely based on their size. Unlike chromatography, this process is effective for all types of proteins, including insoluble proteins or those which are part of large complex aggregates [3]. The disadvantage is that only small quantities of a solution can be used in electrophoresis, as opposed to the large sample volumes that column chromatography allows.
Two-Dimensional Gel Electrophoresis
As one can imagine, proteins that vary by only a few kilobases are very difficult to separate by SDS-PAGE. Even when the starting solution contains thousands of proteins, both column chromatography and SDS-PAGE are only capable of segregating about 50 different proteins at once. To rectify this, the method of two-dimensional (2D) gel electrophoresis was developed, which is capable of resolving around 2,000 different proteins [3]. In the first part of this technique, the individual charges on the proteins are exploited by a process known as isoelectric focusing [24, 25] (Fig. 7.1c). The theory behind this process involves the principle that, since a number of amino acids act as a weak acid or base, proteins can behave as zwitterions (or molecules which can contain both positive and negative charges). Changes in the pH of a protein-containing solution can therefore make acidic or basic amino acid side chains negative, positive, or neutral based on the loss or gain of H+ ions. As a result, all proteins have a characteristic isoelectric point, which is the pH at which the net charge on the entire protein is zero. In other words, proteins will not move within an electrical field when at their isoelectric point. In isoelectric focusing, a pH gradient is first created across a narrow strip of polyacrylamide gel matrix. Proteins are then allowed to move through this gradient in the presence of an electrical field. Each protein will move down the gel until it reaches the pH corresponding to its isoelectric point, then stop as it is no longer charged.
The second dimension of 2D gel electrophoresis relies on the same concepts as SDS-PAGE. After isoelectric focusing, the entire gel is subjected to SDS, which will again bind to and confer a net negative charge on all proteins. The narrow piece of gel is then placed along the edge of a new, standard piece of polyacrylamide gel at a 90° angle, and an electrical field is then applied. As with SDS-PAGE, the proteins will then migrate through the gel according to their size alone. The resulting gel thus contains proteins separated by isoelectric point along its width and by size along its length. As it is extremely unusual for two proteins to possess a similar size and isoelectric point, one may assume that the individual spots on a 2D gel likely contain one unique protein [26].
Historically, criticisms of this technique mostly involved the ability to reliably duplicate each experiment. However, refinements on the products themselves have significantly improved the accuracy of 2D gel electrophoresis. In addition, newer modifications of the procedure, such as the use of fluorescent-tagged proteins to differentially label samples from separate treatment groups which can be run on the same gel (called 2D differential in-gel electrophoresis, or 2D-DIGE) [26, 27], have significantly expanded the value of this method.
Protein Identification and Quantitation
The methods discussed in this section are not only useful to identify and quantify the protein once it has been purified, but they may also be employed as the initial step to one’s study. For example, measuring carcinoembryonic antigen (CEA) in the blood of patients undergoing surveillance for recurrent cancer [28] or measuring other biomarkers of disease such as lipase for pancreatitis [29] and troponin I during a myocardial infarction [30] does not necessarily require protein extraction or purification. Determining which technique to employ to identify the protein of study depends not only on the scientific question but also on the source of the protein and what reagents are available.
Mass Spectrometry and MALDI-TOF
One of the most accurate methods of determining the identity and elemental composition of a protein is through mass spectrometry (MS). In this process, the sample is first loaded into the machine, where it is vaporized then split into separate ions by bombarding them with electrons. The ions are then passed through an electromagnetic field, which bends the fragments into a beam. The speed at which the ions travel depends on both their mass and their charge. The ions are then analyzed and quantified as they pass through the detector. The resulting output is a spectrum of ionic masses unique to each protein [25, 27, 31].
There are a wide variety of methods that use permutations on traditional MS to improve the accuracy of the test. Most commonly used is called matrix–assisted laser desorption ionization time–of–flight spectrometry (or MALDI-TOF) [27, 32]. With this technique, proteins are first cleaved into separate peptide fragments and dried onto a metal or ceramic slide. A laser is then used to ionize the fragments—which is a more gentle process than bombardment with electrons—and the fragments accelerate through the electromagnetic field. With this method, the ionized fragments are analyzed not only by their mass and charge but also by the time it takes to reach the detector, generating a more precise and unique mass spectrum. Tandem mass spectrometers—which sequentially fragment the proteins into peptides, then into individual amino acids—are also used to determine protein sequences. Furthermore, chromatographic methods may be linked to the mass spectrometers to purify and separate the analyzed proteins before they pass through the ionizer.
The main use of MS is for protein identification. Direct sequencing can be performed but it is more cumbersome. More recently, as the field of proteomics is thriving, many are gaining interest in MS again, as this has shown to be useful in determining all posttranslational modifications of a single protein. While MS is perhaps one of the most accurate ways to identify proteins, the process often requires complex analyses using expensive machinery and requires highly purified protein [31].
Western Blotting
As discussed above, SDS-PAGE and 2D gel electrophoresis are reliable methods to separate proteins for further analysis. However, these procedures require additional steps to identify the protein in question. One method involves cutting out the band in question, dissolving the gel, and analyzing the protein by MS [32]. The accuracy of this is quite low, however, as the exact margins of the bands are not always clear and some may contain more than one protein. One of the most reliable ways of detecting and quantifying the protein of study after SDS-PAGE or 2D gel electrophoresis is by Western blotting. Clinical uses of Western blotting include testing for HIV and Lyme disease [33, 34].
With this technique, all the proteins within the gel are transferred to a membrane, typically made from nitrocellulose or polyvinylidene fluoride (or PVDF), both of which nonspecifically bind proteins [3]. Again, all proteins are all negatively charged from the SDS so that, when an electrical field is applied, the proteins will move toward the positive electrode. If a membrane is placed between the gel and the positive electrode (as a “sandwich”), the proteins will transfer to the membrane in a process called electroblotting [3] (Fig. 7.2a).
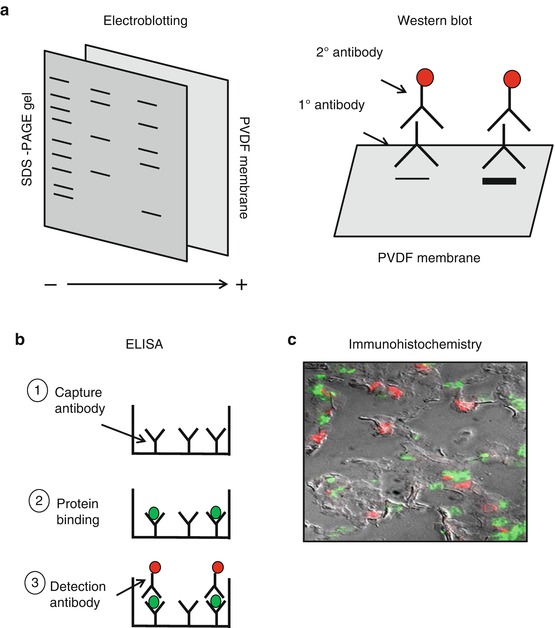
Fig. 7.2
Western blotting and immunoassays. After running protein samples on SDS-PAGE, Western blotting can be used to detect and quantify the protein of interest. As shown in (a), the SDS-PAGE gel is placed on top of a PVDF membrane that nonspecifically binds proteins. Electricity is then applied. The proteins are still negative from the SDS, so they will move toward the positive electrode. Once the proteins are transferred, the membrane is blocked to avoid nonspecific binding of antibodies and then incubated with a primary antibody against the protein being studied. The membrane is then washed and incubated with a labeled secondary antibody. When analyzed, the amount of protein from each unknown sample can be quantified by assessing the thickness of the band on the membrane. (b) To quantify a protein directly in solution, an ELISA can be performed. First, a 96-well plate is coated with a capture antibody that is specific against the protein being studied (1). Next, an unknown sample solution is incubated in the 96-well plate and allowed to bind with the capture antibody (2). The samples are then washed and the detection antibody is added (3). Typically, this antibody is linked to a fluorescent tag or an enzyme that generates a colorimetric change. The amount of the protein of interest in the unknown samples is therefore determined by the intensity of the color or fluorescence, as measured by a spectrophotometer or fluorometer. To determine the concentration of a protein in situ within tissue sections, immunohistochemistry can be applied. This method employs the same methods as other immunoassays and either direct or indirect antibody detection methods can be used. An example of an immunofluorescence image is shown in (c). Here, a section of lung tissue is incubated with a fluorescent-labeled antibody to a neutrophil-specific antibody (green cells). The tissue section is then washed and incubated with a fluorescent-labeled, macrophage-specific antibody (red cells). The total number of neutrophils and macrophages within the lung tissue can then be quantified. To better define the alveolar structure, this image is shown with an overlay of delay interference contrast microscopy (Image courtesy of Dr. Vanessa Nomellini)
The desired protein is then detected and quantified on the membrane using antibodies tagged with a reporter system, typically by a color change or fluorescence (Fig. 7.2b). Since antibodies are proteins themselves, the membrane must first be blocked to minimize nonspecific binding of the antibody. The blocking step is typically performed with a solution containing a small amount of protein, such as bovine serum albumin or evaporated milk. The antibody detection step can be performed in either one step (direct detection) or two steps (indirect detection). If a primary antibody directly linked to a colorimetric or fluorescent tag is not commercially available, the detection must be performed in two steps. For the first step, the membrane is incubated with a solution containing a primary antibody specific to the desired protein derived from an animal other than human. After rinsing off any excess primary antibody, the membrane is then incubated with a solution containing a secondary antibody, typically against the fragment crystallizable (Fc) portion of the primary antibody. For example, if the primary antibody was made in a goat against a human antigen, the secondary antibody would have to be against the Fc portion of goat antibodies. There are a wide variety of secondary antibodies available that are tagged with either a fluorescent label or a reporter enzyme that generates a colorimetric or chemiluminescent by-product when combined with its substrate. A commonly used enzyme reporter system is horseradish peroxidase, which has a number of substrates and can generate either colored or fluorescent by-products. Another system utilizes the strong affinity between biotin and streptavidin. If the antibody is constructed with a biotin molecule attached, this can be incubated with streptavidin molecules coated with a fluorescent dye. With the appropriate device, such as a spectrophotometer or a fluorometer, the intensity of the color or fluorescent signal can be detected and quantified relative to a standard curve with known concentrations of protein.
Although Western blotting has a high degree of sensitivity and specificity, there are a number of limitations to this method. First, the electroblotting aspect of this technique does not guarantee that 100 % of all proteins within the SDS-PAGE gel will be transferred to the membrane or that all transferred proteins will actually bind to the membrane. In terms of using one- or two-step antibody binding, there are a number of advantages and disadvantages. In the direct detection method, the process is quicker since only one antibody step is required. Indirect methods require the use of more than one antibody, which not only increases the time of the procedure but also increases the risk of cross-reactivity of the protein with other proteins on the membrane. In the direct method, however, the antibody itself is labeled, potentially affecting the binding affinity with its target, which is not the case with indirect detection methods. Furthermore, antibodies can only be constructed with a limited number of enzyme or fluorescent tags, making little opportunity for signal amplification. With the indirect method, multiple tagged secondary antibodies can bind to the primary antibody, which allows for signal amplification and detection of very low concentrations of proteins [35].
Immunoassays
The principles of immunoassays are similar to those used for Western blots. The main difference is that the procedure does not require the separation of a protein solution by SDS-PAGE. There are a number of immunoassays currently in use, including enzyme-linked immunosorbent assay (ELISA), multiplex assays, and enzyme-linked immunospot assays (ELISpot).
As opposed to immobilizing proteins on a membrane for antibody detection, the ELISA (Fig. 7.2c) allows for a protein-containing solution to be measured directly in solution within a 96-well plate. Most uncoated, plastic, 96-well plates will nonspecifically bind a majority of proteins, including either the protein of interest or antibodies. If the starting solution is a purified protein, an ELISA may be performed via a direct method whereby the protein is directly adsorbed onto the wells and quantified using labeled antibodies. If starting with an unpurified protein mixture, such as that from a tissue homogenate or cell lysate, an indirect (or “sandwich”) method is typically employed. Here, the primary antibody itself (called the capture antibody) is first adsorbed onto the wells of the plate. The antibodies are then exposed to the protein mixture and selectively bind the targeted protein. Ideally, there are more capture antibodies coating the plate than the amount of the target protein in the unknown solution. If this is not the case, the protein solution can be diluted until one can ensure that all the protein in the sample will bind to the capture antibody and not over-saturate the system. The concentration of the protein is then determined by a subsequent antibody system (the detection antibody), either using another primary antibody directly tagged with an enzyme or an unlabeled primary and tagged secondary antibody as described above. The amount of protein present is then determined by adding the enzyme substrate and quantifying the resulting signal [36, 37]. For some common proteins, pre-made plates can be purchased which are already coated with the capture antibody of interest, making the process even quicker.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


