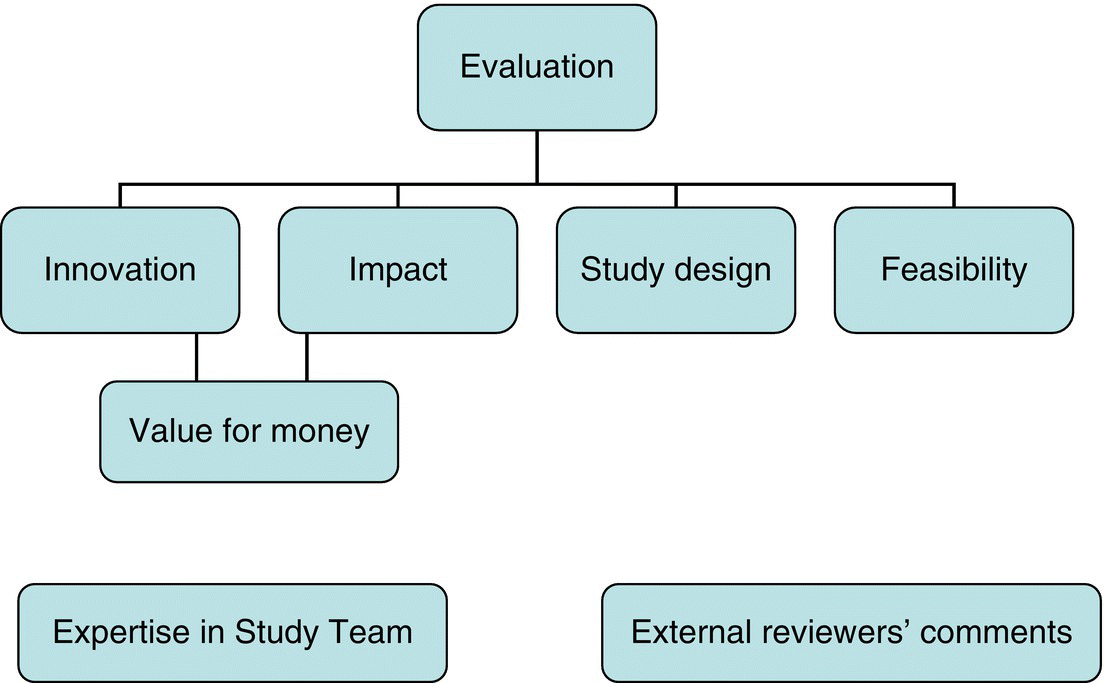
Chapter 11 There are established systems for conducting and reporting observational studies, and most will proceed through a standard set of stages (Box 11.1). A small multidisciplinary working group of key people (perhaps three to five, but possibly more) should initially develop the project. The group could include relevant health professionals, a statistician, and other members with specialist backgrounds. After securing funding, the group can expand to form the study team (sometimes also called study management group, or study steering group/committee) to manage the project over its entire duration. This team could additionally include expertise in data management, regulations and safety monitoring, IT (database systems). It is also worth including potential collaborators, especially recruiting centres from which either participants or data will be obtained. The cost of conducting an observational study varies; small-scale studies, which only involve retrieving data from medical records within a single department or institution, may not incur costs if the researcher is already a member of staff and conducts the project as part of their work. However, multicentre studies, in which participants are recruited and followed up for several months or years, can be expensive to set up and conduct and so require staff to co-ordinate the project. The number and type of staff required will depend on the complexity of the study and sample size, and could include staff to perform and provide any of the following: The salary costs for these staff will depend on the time they are expected to spend on the study and where the work will be undertaken (central research unit or recruitment at centres). Other direct costs to consider include: Grants to conduct observational studies are limited and competitive, and they usually come from governmental bodies, research councils, charities, or private benefactors. Pharmaceutical companies do not often provide grants for observational studies. However, many academic departments are able to run small- or even moderate-sized observational studies without external funding, and studies based on data already collected should require minimal resources, if any (perhaps only for the statistical analysis and some IT input). The previous section covered direct costs attributed to the study, but for institutions such as universities, there can be overheads (indirect costs) that need to be added to the grant. These costs are meant to cover central support and administrative resources, used by staff conducting the study who are employed by the institution. Figure 11.1 shows the main items to address in a typical grant application. Funding bodies seek value for money and are likely to only support studies that have the potential to change practice or that involve novel translational research. If funding is to be sought, the key issues of the application should be considered thoroughly by the working group to ensure that there are no oversights or errors that could be picked up for the first time by the funding committee or their external reviewers. Although the format of the application forms will vary between funding organisations, many aspects are often covered by the study protocol1 (see page 219). Further details about how to develop grant applications can be found elsewhere [1]. Figure 11.1 Key aspects of a typical grant application used by a funding committee for evaluation purposes. Some research departments have the primary purpose to design, set up, and analyse studies involving humans, and so should have permanent staff in place, often including clinicians, health professionals, statisticians, co-ordinators, data managers, and IT/database staff. Having access to such departments is important for large-scale studies, particularly if they involve following up the study participants. Potential recruiting centres, sometimes referred to as a site, should be identified, with a realistic estimate of the number of expected participants per site (investigators tend to overestimate this). This helps ensure that the target sample size is feasible in a reasonable time frame. A sponsor is the institution with ultimate responsibility for the design and conduct of the study. The concept of a sponsor is often only used for studies that involve recruiting participants and obtaining information (including biological samples) directly from them, and also when they are to be followed up for several months or years. Studies using data already collected probably do not need a sponsor, but local guidelines should be checked on this. The chief investigator is the lead researcher for the study and often first developed the idea. A Principal Investigator (PI) is an individual responsible for the study at a single recruiting centre, so a multicentre study would have several PIs. These are not standard labels, but their roles often are. A chief investigator is sometimes called a PI. A protocol should be developed for any study that involves recruiting participants. It may be required as part of a grant application submission, otherwise it must be developed before application for ethical approval is made. It provides a summary of the justification for the study, details of the design, and a set of instructions for sites and (if applicable) the co-ordinating centre, describing how participants are to be recruited (including eligibility) and followed up. It should provide the basis to ensure that the study is conducted to a similar standard across all sites. Studies that do not involve direct contact with participants (e.g. those only using data to be extracted from medical records) might still benefit from having a (usually much shorter) protocol, focussing on the study design and analysis. The protocol can be any length, as long as it contains a clear plan of what will happen to participants, from the time they consent to the time they leave the study. Box 11.2 shows suggested key sections in a protocol. An important part of the protocol is describing how biological samples should be collected, processed, and stored. If this is not specified in the protocol, there should be an Standard Operating Procedure that provides these details (see page 231). All participants are required to give informed consent before taking part for many observational studies. This includes studies where people are: Depending on local regulations and guidelines, there are studies in which informed consent may not be required, such as those using only data from medical records, or stored biological samples (often linked with clinical data). Indeed, there is evidence that if informed consent were obtained in studies like these, the characteristics of those who agree to participate differ from those who do not, creating a bias in the study outcome measures and results [2]. Also, there are some studies where obtaining consent from each participant is not possible or feasible. In others, where people are asked to complete a questionnaire, those who do so have implicitly consented. In these situations, although participants are not asked directly to take part, there should still be an independent review and approval of the project by an independent ethics committee (see page 225), who will stipulate whether or not participants’ consent is required. When informed consent is required, sufficient information about the study must be provided to allow potential participants to understand the objectives and examine the possible benefits and risks of taking part. Information may be provided verbally, but it is usually given as a leaflet: the Participant Information Sheet (PIS). After reading this, participants sign and date a consent form, which is co-signed by an authorised staff member. Suggested sections are shown in Boxes 11.3 and 11.4. Signed consent forms should be kept in the site files, and a copy given to the participant. Sometimes, signed forms are not appropriate, for example, studies of sensitive issues such as taking illicit drugs or sexual habits, because anonymity is needed. Consent may also be obtained by telephone, if this is how data are to be collected anyway. If a potential participant is unable to provide informed consent, for example, children or incapacitated adults, a guardian or representative (usually a parent, or other close relative) could be asked instead. This must be made clear in the application for independent ethics review. Anonymity is an important feature of research (i.e. individual participants cannot be identified by researchers, nor should they be identifiable in publications), and it should be specified in the PIS. Participant names or initial with dates of birth should not be used routinely. However, if the researchers require person identifiers (e.g. full name, address, national identification numbers, initials, and date of birth), this must be specifically requested on the consent form (Box 11.4). For example, there are studies with regular questionnaires (e.g. yearly) to be posted to participants, so full contact details are essential to the study. There are often clear local guidelines or regulations regarding data protection and confidentiality. The text in the PIS and consent form should be written in simple language. It is often useful to ask a few potential participants, lay people, members of a patient representative group or research nurse, to comment on the text before it is finalised, particularly for complex studies. This is because they may read and interpret the PIS differently from the researchers. There are studies in which a previously undiagnosed disorder is found in a participant. If this disorder is among the study objectives, then it is appropriate that the result is sent to the participant and his/her family physician, and the procedures for this should be specified in the protocol and the PIS. If the disorder is unrelated to the study objectives (i.e. an incidental finding), the issue requires consideration, particularly when it is one that should be treated. Further investigations, with possible interventions, would not be part of the study protocol, but if the disorder can be readily linked to the participant, then it is appropriate to inform them, usually through their family physician, after which they can seek standard care outside of the study. Genetic testing using donated biological specimens is becoming more common. It may be done among healthy participants (e.g. general population) or those who already have a disorder (e.g. cancer patients). However, there are potential issues with this regarding what information, if any, is reported back to participants. Finding a ‘high risk’ gene or genetic mutation is possible, but the researchers must carefully consider whether it is one that is established to be correlated with a disorder (or other outcomes such as early death). There will be many mutations for which the clinical implications are as yet unknown, and there may even be consequences for health insurance. Offering genetic results to participant’s (in prospective studies) may be recommended, when all of the following four conditions hold [3]: If the genetic results can be readily linked to individuals, or the participants expect to be told the results, the researchers must have a system in place for reporting this information, counselling and appropriate interventions for those found to have an abnormal result. All of this will probably require funding. Furthermore, while some genetic testing may be done during or at the end of the study, some studies will only perform this in the future, so there will be ethical implications in contacting a study participant from several/many years before, regarding a detected genetic defect or high-risk gene. Finally, it is important that if genetic results are to be reported back to participants, this should be specified in both the PIS and consent form, that is, the participants need to agree to this. If genetic testing is to be kept anonymous, that is, participants are not informed, this should also be stated (example in Box 11.5).
Conducting and reporting observational studies
11.1 Before conducting the study
Establishing a working group and a study team
Estimate the financial costs of the study
Grant funding
11.2 Study set-up
Sponsor, lead study researcher, and other investigators
Study protocol
Informed consent, participant information sheet, and consent form
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


