Chapter 14 Richard O. Francis; Jeffrey S. Jhang B. Type 3. C. Type 2B. D. Type 2A. E. Type 2M. 2. A 2-month-old boy with a history of gut malrotation is scheduled to undergo corrective surgery. Preoperative laboratory tests include coagulation screening tests as shown in Table 14-1. The surgery is performed without any bleeding complications. Which one of the following clotting factors has similar or higher activity levels in younger children than in adults? B. Factor II. C. Factor X. D. Factor XI. E. Prekallikrein. 3. A 4-year-old girl with epistaxis and significant bleeding with loss of her baby teeth is brought to her pediatrician by her mother. The patient had a laboratory evaluation, including an evaluation for vWD. The relevant laboratory findings are shown in Table 14-2. A ristocetin-induced platelet aggregation study was performed (Figure 14-1). Which one of the following steps would be most useful in distinguishing the disorders that could account for these findings? A. Desmopressin (DDAVP) challenge. B. Cryoprecipitate challenge. C. Platelet function analyzer (PFA)-100. D. Partial thromboplastin time (PTT) mixing study. E. von Willebrand factor (vWF) multimer analysis. 4a. A 20-year-old woman with a history of menorrhagia was recently diagnosed with vWD. Her laboratory results are shown in Table 14-3. Which one of the following types of vWD is most consistent with the laboratory values? B. Type 2N. C. Type 2A. D. Type 2M. E. Type 1. 4b. The initial diagnosis of the patient was delayed because vWF antigen and activity results were within the reference range when she was initially evaluated 2 years earlier. In the interim, however, she continued to have menorrhagia, prompting reevaluation and the subsequent diagnosis of type of vWD in Question 4a. Which one of the following is the most likely reason that vWD was not diagnosed on her initial visit? B. At the time of initial evaluation the patient was taking oral contraceptives, which are known to increase vWF levels. C. The blood sample for testing was transported to the laboratory at room temperature instead of the preferred temperature of 4°C. D. At the time of the initial evaluation, the patient had not recently exercised, and a lack of physical activity is known to increase vWF levels. E. vWF levels are known to decrease with increasing age. 5. Which one of the following is an acquired cause of factor VII deficiency? A. Left ventricular assist device. B. Inflammatory bowel disease. C. Phytonadione therapy. D. Heparin therapy. E. Ganciclovir therapy. 6. Which one of the following clotting factors has the longest plasma half-life? B. Factor X. C. Factor II. D. Factor XIII. E. Factor VIII. 7. Which one of the following is a rare complication of congenital factor VII deficiency? B. Thrombosis. C. Menorrhagia. D. Central nervous system bleeding. E. Postoperative bleeding. 8. A 3-day-old infant, born by normal spontaneous delivery without complications, is brought to the emergency department with vomiting, lethargy, and pallor. On physical examination, the anterior fontanelle is bulging, and a cephalohematoma is diagnosed. A complete blood cell count shows a hemoglobin of 7.2 g/dL (11.0 to 13.0 g/dL), platelet count of 254 (165 to 415 × 109/L), prothrombin time (PT) of 62.4 seconds (12.3 to 14.6 seconds), and activated partial thromboplastin time (aPTT) of 32.1 seconds (24.0 to 34.6 seconds). The PT corrects into the reference range when mixed 1:1 with pooled normal plasma. Which one of the following factors is most likely to be decreased in this patient? B. Factor VII. C. Factor VIII. D. Factor IX. E. Factor XI. 9. Which one of the following reasons makes it difficult to treat actively bleeding patients with congenital factor VII deficiency using recombinant factor VIIa? A. Formation of anti–factor VII antibodies. B. Short half-life of recombinant factor VIIa. C. Requirement for high doses (120 μg/kg). D. Contamination with activated factors II, IX, and X. E. Need to overlap with heparin therapy. 10a. A 28-year-old man with known hemophilia A is seen in the emergency department at 3 am with hemarthrosis after getting into a fight at a local bar. He occasionally develops hemarthrosis and has received factor replacement therapy. The patient’s factor VIII activity is reported as less than 1%. The patient weighs 100 kg, and 2500 IU of factor VIII concentrates was administered. The following morning, a right forearm muscle hematoma was expanding and the affected joint was not improving. A factor VIII level 12 hours after administration was 4%. A 1:1 mixing study and a 2-hour incubated mixing study were performed. Which one of the following is the best explanation for the clinical and laboratory findings shown in Table 14-4? A. Consistent with factor VIII half-life. B. Consistent with inadequate dosing of factor VIII. C. Inadequate reconstitution of lyophilized product. D. Presence of a lupus anticoagulant (LAC). E. Factor VIII inhibitor. Table 14-4 * Activated partial thromboplastin time in seconds. 10b. The desired target for factor VIII activity was 50% in this patient. Which one of the following is the correct initial dose of factor VIII concentrate that should be administered? B. 1000 IU. C. 2500 IU. D. 3500 IU. E. 5000 IU. 11a. An 8-year-old boy presents to the emergency department with a left knee hemarthrosis and persistent bleeding from a cut after falling while playing tennis. The parents were told that their child has hemophilia but they do not know what type. He has never had any bleeding and has not received any clotting factors. His coagulation studies are shown in Table 14-5, and a 1:1 mixing study with pooled normal plasma shows correction of the aPTT into the reference range. Which one of the following is the most likely factor that is deficient in this patient? B. Factor VII. C. Factor VIII. D. Factor X. E. Factor V. 11b. The patient has never been transfused and has not received factor replacement therapy. A factor IX activity is reported as less than 1% and factor VIII as 82%. The patient weighs 30 kg, and a target of 50% factor IX activity is desired. Which one of the following is the appropriate dose of recombinant factor IX replacement? B. 210 IU. C. 750 IU. D. 1500 IU. E. 5000 IU. 12. What is the biologic half-life of factor IX? B. 12 hours. C. 24 hours. D. 48 hours. E. 96 hours. 13. Which one of the following clinical manifestations is characteristic of patients with a coagulation factor deficiency? B. Superficial hematomas. C. Epistaxis. D. Delayed bleeding. E. Gum bleeding. 14. Which one of the following coagulation factor proteins is vitamin K dependent? B. Factor II. C. Factor VIII. D. Factor V. E. Factor XIII. 15. Polymorphisms in which one of the following genes is associated with variable response to warfarin therapy? B. CYP2D9. C. ABCB1. D. CYP3A4. E. CYP2J2. 16. A 32-year-old Ashkenazi Jewish woman, 38 weeks pregnant (G2P1001), is in active labor and is failing to progress. A cesarean delivery is planned. During the delivery of her first child, she bled profusely and required transfusion of 2 U of packed red cells. At her obstetrician’s office, records show that she has a complete blood cell count with a hemoglobin of 10.2 g/dL (11.0 to 13.0 g/dL), platelets 254 (165 to 415 × 109/L), PT 13.4 seconds (12.3 to 14.6 seconds), and aPTT 54.1 seconds (24.0 to 34.6 seconds). The patient’s aPTT corrected into the reference range when mixed 1:1 with pooled normal plasma. Factor levels are factor VIII 180% (50% to 150%), factor IX 107% (50% to 150%), and factor XI 12% (50% to 150%). Which one of the following is the best choice to treat active bleeding in this patient? A. Recombinant activated factor VII, 90 μg/kg. B. Fresh frozen plasma (FFP), 15 mL/kg. C. Recombinant factor VIII. D. Fibrin glue. E. Recombinant factor XI. 17. Which one of the following coagulation protein deficiencies is not associated with a bleeding disorder? B. Factor VII. C. Factor XI. D. Factor XII. E. Factor XIII. 18. A 73-year-old man’s coagulation studies are shown in Table 14-6. A 1:1 mixing study corrected the patient’s plasma aPTT into the reference range. When the incubation time of the patient plasma with the aPTT reagent was increased to 10 minutes, the aPTT decreased to 30.6 seconds. Which one of the following disorders is consistent with these findings? B. Protein C deficiency. C. Factor XIII deficiency. D. Prekallikrein deficiency. E. High-molecular-weight kininogen deficiency. 19. Your laboratory performs a factor X activity assay using a minimum of three dilutions of patient plasma. The three plasma dilutions exceed the allowable within-run coefficient of variation. Which one of the following disclaimers should be reported with the result? B. Inhibitor pattern. C. Noncalibrated curve. D. Linear range. E. Between-subject variability. 20. Which one of the following acquired coagulation factor deficiencies is associated with primary systemic amyloidosis? B. Factor VII. C. Factor VIII. D. Factor X. E. Factor XII. 21. A 5-day-old infant has delayed bleeding from the umbilical cord stump. Laboratory studies are shown in Table 14-7. A 5 M urea test is performed on the patient’s sample and shows rapid dissolution of the clot (decreased clot stability). Which one of the following disorders is the most likely to be the cause of bleeding? B. Dysfibrinogenemia. C. Factor XIII deficiency. D. Fibrinolysis. E. High-molecular-weight kininogen deficiency. 22. Your laboratory is validating a new aPTT reagent. To determine the heparin therapeutic range, samples from heparinized patients are tested with the aPTT reagent and then an anti-Xa assay is performed. The linear regression line is shown in Figure 14-2. Which one of the following is the best therapeutic range according to this Brill-Edwards experiment? B. 40 to 60 seconds. C. 60 to 80 seconds. D. 60 to 120 seconds. E. 80 to 120 seconds. 23. For which one of the following clinical situations would thrombophilia testing be most indicated? A. 45-year-old man with a second cousin who had a venous thrombosis. B. 49-year-old man with hypertension, hyperlipidemia, and diabetes with first stroke. C. 55-year-old man with first mesenteric vein thrombosis. D. 35-year-old woman with first spontaneous abortion in first trimester. E. 45-year-old woman taking oral contraceptives. 24. Which of the following can lead to falsely low protein S activity on a PTT-based protein S activity assay? A. Decreased factor VIII activity. B. Decreased factor V activity. C. Direct thrombin inhibitor. D. Factor V Leiden mutation. E. Pregnancy. 25a. A 29-year-old woman who has had three consecutive spontaneous abortions over the past 2 years presents to her physician complaining of pain, swelling, and redness in her left leg. Ultrasound of her lower extremities reveals a deep venous thrombosis (DVT) in her left leg. She has no history of prior DVT. Her complete blood cell count is normal. Coagulation study results are shown in Table 14-8. Which one of the following is the most likely cause of the prolongation of the aPTT? B. Severe factor VII deficiency. C. LAC. D. Disseminated intravascular coagulation (DIC). E. Factor IX inhibitor. 25b. Which one of the following must be demonstrated during laboratory testing to identify an LAC? A. Phospholipid independence of the inhibitor. B. Phospholipid dependence of the inhibitor. C. Mixing study in which the aPTT corrects into the reference range. D. Prolongation of a phospholipid-independent clotting test. E. Mixing study where the aPTT prolongation is time dependent. 25c. Which one of the following is an antiphospholipid antibody that is evaluated for the diagnosis of antiphospholipid antibody syndrome? A. Anti–glomerular basement membrane (anti-GBM) antibody. B. Antinuclear antibody. C. Anti–Jo-1 antibody. D. Anti–ADAMTS-13 antibodies. E. Anti–β2-glycoprotein 1 antibodies. 25d. The patient is tested for the presence of an anticardiolipin (aCL) antibody following baseline coagulation studies (Table 14-9). A high-titer aCL antibody is detected. Which one of the following best describes whether the patient meets the criteria for the diagnosis of antiphospholipid antibody syndrome (APS) at this time? B. A DVT and positive mixing study showing the presence of an inhibitor are diagnostic for APS. C. An arterial thrombosis is necessary to diagnose APS. D. An antiphospholipid antibody must be demonstrated on more than one occasion to meet the diagnostic criteria for APS. E. Detection of any antiphospholipid antibody is itself diagnostic of APS. 26. Which one of the following is the best therapy to treat the findings shown in Figure 14-3, B? B. Aspirin. C. Packed red blood cell transfusion. D. Plasma transfusion. E. Platelet transfusion. 27. Which one of the following is the best therapy to treat the findings shown in Figure 14-4, B? B. Heparin. C. Packed red blood cell transfusion. D. Plasma transfusion. E. Platelet transfusion. 28. Which one of the following is the best therapy to treat the findings shown in Figure 14-5, B? B. Heparin. C. Packed red blood cell transfusion. D. Plasma transfusion. E. Platelet transfusion. 29. Which one of the following assays is the best method for measuring antithrombin (AT) activity? B. Enzyme-linked immunoassay (ELISA). C. Latex particle immunoturbidometric. D. DNA analysis. E. Heparin-binding site inhibition. 30. Which one of the following tests is the most sensitive in detecting dysfibrinogenemia? A. Antigenic-based fibrinogen. B. Fibrometer-based fibrinogen. C. Nephelometry. D. Reptilase time. E. Thrombin time. 31. Which one of the following is the closest approximation of the prevalence of the factor V Leiden mutation in Caucasian Americans? B. 1%. C. 5%. D. 15%. E. 25%. 32. Which one of the following is an acquired cause of low AT activity? A. Increased heparin cofactor II. B. l-Asparaginase therapy. C. Hematuria. D. Menopause. E. Warfarin therapy. 33. Your laboratory performs a second-generation modified functional activated protein C (APC) resistance screen by determining the activated protein C resistance (APCR) ratio of an aPTT. Which one of the following situations will most likely cause a false-positive result? B. Warfarin therapy. C. Heparin 0.7 U/mL. D. LAC. E. Liver dysfunction. 34. Which one of the following can increase protein C activity in an aPTT-based assay? A. Elevated factor VIII activity. B. Factor V Leiden. C. LAC. D. Warfarin therapy. E. Thrombosis. 35. Which one of the following disorders can present with purpura fulminans and DIC in newborns? A. Double heterozygous protein S and AT deficiency. B. Dysfibrinogenemia. C. Homozygous factor V Leiden. D. Homozygous protein C deficiency. E. Homozygous prothrombin 20210G>A mutation. 36. Which one of the following is an effect associated with heparin therapy? B. Development of anti–heparin polyvinyl sulfate antibodies. C. Development of Hollenhorst plaques. D. Hypokalemia. E. Hirsutism. 37. The international sensitivity index (ISI) of the thromboplastin used in your laboratory is 1.26 according to the package insert. You perform the PT with this reagent in 120 normal donors and find the arithmetic mean to be 13.5 seconds, the geometric mean 13.7 seconds, and the standard error 3.5%. A patient’s PT is 19.8 seconds using this reagent. Which one of the following is the closest value for the international normalized ratio (INR) for this patient? (Note: A calculator to determine the value is allowed.) B. 1.47 C. 1.59 D. 1.62 E. 5.67 38. A 64-year-old man develops a DVT after a right total hip replacement. Before initiating heparin therapy, laboratory studies show a PT of 14.8 seconds (12.7 to 15.4) and an aPTT of 29.3 seconds (26.3 to 39.4). The heparin therapeutic range is an aPTT of 60 to 80 seconds. Heparin therapy is initiated with a bolus of 5000 IU and then 1000 IU/hr. Six hours later, the aPTT is 35 seconds. Another bolus of 5000 IU of heparin is administered, and the infusion is increased to 1500 IU/hr. The aPTT 6 hours later is 44 seconds. An anti-Xa level is performed and found to be 1.1 IU/mL. Which one of the following is the most likely explanation for these findings? B. AT deficiency. C. Acquired factor VIII deficiency. D. APCR. E. Increased heparin cofactor II activity. 39. A 55-year-old man requires heparin reversal after cardiopulmonary bypass for aortic valve repair. Which one of the following is the best method for reversal of the anticoagulant effect of unfractionated heparin (UFH) in this patient? B. Vitamin K. C. Cryoprecipitate. D. Prothrombin complex concentrates. E. Protamine sulfate. 40. The PT for 13 normal donors and patients receiving stable warfarin therapy is tested with a reference thromboplastin and your new thromboplastin preparation (test reagent). A double logarithmic plot of the PT (seconds) of the reference preparation versus the test thromboplastin reagent is shown along with the regression equation (Figure 14-6). If the ISI of the reference preparation is 1.0, which one of the following is the best estimation of the ISI of the test reagent? B. 0.90 C. 0.95 D. 1.0 E. 1.1 41. Which one of the following is an adverse effect of warfarin therapy? B. Transmissible spongiform encephalopathy. C. Purple toe syndrome. D. Immune thrombocytopenia. E. Hollenhorst plaque. 42. Several hemostatic changes occur during the different stages of the liver transplant procedure. Which one of the following stages of liver transplantation is associated with hypercoagulability? B. Anhepatic to reperfusion. C. Postreperfusion. D. Postoperative. E. During surgical dissection and mobilization of the diseased liver. 43. Which one of the following is the mechanism of thrombosis associated with AT III deficiency? A. Endothelial cell cytotoxicity and disturbance of vascular hemostatic mechanisms. B. Failure to generate APC. C. Failure of APC to inactivate factors Va and VIIIa. D. Failure to generate plasmin. E. Failure to inhibit factors IIa (thrombin), Xa, and other activated factors. 44. Which one of the following is a recognized mechanism for thrombosis in patients with dysfibrinogenemia? A. Abnormal fibrinogen neutralizes tissue plasminogen activator (tPA). B. Abnormal fibrinogen inhibits protein C activity. C. Abnormal fibrinogen resists fibrinolysis. D. Abnormal fibrinogen induces cytotoxicity and disrupts vascular hemostatic mechanisms. E. Abnormal fibrinogen disrupts the activation of plasminogen. 45. Which one of the following is the mechanism of thrombosis in the setting of factor V Leiden? B. Neutralization of tissue plasminogen activator (tPA). C. Endothelial cell toxicity and disruption of vascular hemostatic mechanisms. D. Activated protein C is unable to inactivate factor Va. E. Failure to inhibit thrombin. 46. Which one of the following is the mechanism for thrombosis in the setting of heparin cofactor II deficiency? A. Failure to inhibit thrombin. B. Failure to activate plasminogen. C. Failure to inhibit factor IIa (i.e., thrombin), factor Xa, and other activated factors. D. Failure to generate APC. E. Neutralization of tPA. 47. Which one of the following anticoagulant factors is solely produced by the liver? B. AT. C. Protein S. D. Plasminogen. E. Tissue factor pathway inhibitor. 48. Which one of the following procoagulant hemostatic factors is produced solely by the liver? B. Fibrinogen. C. Factor V. D. Plasminogen activator inhibitor. E. Plasminogen. 49. Which one of the following is the mechanism of thrombosis associated with hyperhomocysteinemia? B. Failure to inhibit factor IIa (i.e. thrombin), factor Xa, and other activated factors. C. Failure to activate plasminogen. D. Endothelial cell toxicity and disruption of vascular hemostatic mechanisms. E. Failure of APC to inactivate factors Va and VIIIa. 50. Which one of the following statements most accurately reflects the utility of routine coagulation tests to predict bleeding or thrombotic risk in liver disease? B. There is good evidence that the PT, not the PTT, is useful in predicting bleeding or thrombotic risk in liver disease. C. There is no good evidence to support the use of routine coagulation tests for predicting bleeding or thrombotic risk in liver disease. D. There is good evidence to support the use of the PTT for predicting bleeding or thrombotic risk in liver disease. E. There is good evidence to support the use of the thrombin time for predicting bleeding or thrombotic risk in liver disease. 51. Which one of the following disorders is associated with neutralization of tPA? B. AT deficiency. C. Factor V Leiden. D. Heparin cofactor II deficiency. E. Excess plasminogen activator inhibitor 1 activity. 52. Which one of the following mechanisms for thrombosis is associated with plasminogen deficiency? A. Failure of APC to inactivate factors Va and VIIIa. B. Failure to inhibit thrombin. C. Failure to generate plasmin. D. Failure to generate APC. E. Failure of activated protein C to inactivate factor Va. 53. Which one of the following is the mechanism of thrombosis in protein C deficiency? A. Failure to inhibit factor IIa, factor Xa, and other activated factors. B. Failure to generate APC. C. Failure to generate plasmin. D. Endothelial cell toxicity and disturbance of vascular hemostatic mechanisms. E. Failure of APC to inactivate factors Va and VIIIa. 54. Which one of the following is the mechanism of thrombosis in protein S deficiency? A. Failure to inhibit thrombin. B. Failure of APC to inactivate factors Va and VIIIa. C. APC fails to inactivate factor Va because of a highly conserved point mutation. D. Failure to activate plasminogen. E. An acquired abnormality in fibrin results in decreased fibrinolysis. 55. Which one of the following is the mechanism of thrombosis associated with tPA deficiency? A. Endothelial cell toxicity and disruption of vascular hemostatic mechanisms. B. Failure of APC to inactivate factors Va and VIIIa. C. Failure to generate APC. D. Failure to activate plasminogen. E. Failure to inhibit factor IIa, factor Xa, and other activated factors. Major points of discussion ■ Types 1 and 3 vWD are characterized by quantitative deficiencies of normal vWF. ■ Type 2A vWD is caused by a mutation in the vWF protease cleavage site, leading to increased enzymatic cleavage and a lack of high- and intermediate-molecular-weight multimers. ■ Type 2B vWD is caused by a gain-of-function-mutation in the platelet GPIb/V/IX binding site of vWF, leading to a “stickier” vWF. ■ Type 2M vWD is caused by a loss-of-function-mutation in the platelet GPIb/V/IX binding site of vWF, leading to decreased binding of vWF to platelets. ■ Type 2N vWD is caused by a mutation in the factor VIII binding site of vWF. ■ Thrombocytopenia following DDAVP administration in type 2B vWD is usually transient and often is not associated with bleeding or thrombosis. ■ DDAVP may be cautiously considered for patients with type 2 vWD.18 2. A. Fibrinogen. Major points of discussion ■ Interpretation of screening tests, as well as clotting factor activities, should be based on age-specific reference intervals. ■ Clotting factors with lower levels in young children than in adults include the vitamin K–dependent factors (II, VII, IX, and X), as well as factors V, XI, and XII, prekallikrein, and high-molecular-weight kininogen. ■ Clotting factors with similar or higher levels in young children than in adults include fibrinogen, factor VIII, and vWF. ■ Because of the age-related changes in clotting factor levels that occur early in life, repeat testing at a later age may be necessary to exclude or confirm a diagnosis of bleeding related to a factor deficiency in a neonate.1 3. A. Desmopressin (DDAVP) challenge. Major points of discussion ■ PT-vWD is caused by a gain-of-function mutation in GPIbα that leads to increased binding to vWF. ■ Type 2B vWD and PT-vWD have similar clinical and laboratory findings. Although variable, patients classically present with thrombocytopenia, decreased vWF:Ag and vWF:ristocetin cofactor activity (RCo), increased vWF:Ag/vWF:RCo ratio, lack of high-molecular-weight vWF multimers, decreased FVIII:C, and abnormal ristocetin-induced platelet aggregation. ■ It is important to distinguish these two disorders because the treatment will differ. Administration of vWF/FVIII concentrates in vWF type 2B would be effective, whereas in PT-vWD, it would not. Desmopressin (DDAVP) is contraindicated in PT-vWD. ■ The cryoprecipitate challenge is a laboratory test that tests spontaneous platelet aggregation after the addition of cryoprecipitate containing normal vWF. The challenge will lead to spontaneous aggregation in PT-vWD, but not in type 2B vWD.9 4a. A. Type 3. 4b. A. The patient is blood type O, and these individuals tend to have higher baseline vWF antigen levels than those with other blood groups. Major points of discussion ■ Several factors affect vWF levels and can confound the diagnosis of vWD. ■ vWF is an acute-phase reactant and therefore is increased by stress, inflammation, pregnancy, and oral contraceptives. Repeated testing is necessary in some cases to identify low vWF levels. ■ Medical conditions that can decrease vWF levels or activity include autoimmune/antibody-induced inhibition or clearance and destruction of vWF by increased shear stress (e.g., left ventricular assist devices). ■ ABO blood type has an effect on plasma vWF levels. Individuals with group O blood type have approximately 25% lower vWF levels than individuals with other blood types. ■ vWF levels tend to be higher in neonates than adults. ■ Approximately 30% of variation in vWF levels is heritable.18 5. A. Left ventricular assist device. Major points of discussion ■ Acquired factor VII deficiency can be due to acquired vitamin K deficiency (e.g., diet, antibiotic therapy, total parenteral nutrition, and gastrointestinal malabsorption), liver disease, and warfarin therapy. ■ Vitamin K is required for the γ-carboxylation of the vitamin K–dependent coagulation factors (factors II, VII, IX, and X and proteins C and S). In the absence of vitamin K and γ-carboxylation, these factors are not active. ■ Recycling vitamin K requires enzymes that are inhibited by warfarin, which leads to inactive vitamin K–dependent coagulation and anticoagulation factors. Exogenous administration of vitamin K (phytonadione) is used as an antidote for the warfarin effect. ■ Antibiotics can alter gut flora, which leads to vitamin K deficiency. Hospitalized patients are often taking antibiotics and may be NPO or receiving total parenteral nutrition, which can make them vitamin K deficient. ■ Hemorrhagic disease of the newborn can be seen in neonates who do not get adequate vitamin K across the placenta before delivery. ■ The vitamin K–dependent factors II, VII, IX, and X and proteins C and S are produced in the liver. Decreased synthetic ability of the liver leads to decreased coagulation factor levels. 6. A. Factor VII. Major points of discussion ■ For example, recombinant factor VIIa is dosed every 2 hours, factor VIII every 12 hours, and factor IX every 24 hours. ■ Factor VII has the shortest half-life. ■ Factor XIII has the longest half-life. ■ The approximate plasma half-lives of the clotting factors are as follows: • Factor II: 65 hours • Factor V: 15 to 36 hours • Factor VII: 5 hours • Factor VIII: 10 hours • Factor IX: 25 hours • Factor X: 40 hours • Factor XI: 45 to 80 hours • Factor XII: 50 to 70 hours • Factor XIII: 200 hours 7. A. Epistaxis. Major points of discussion ■ It is an autosomal recessive disorder and has an incidence of 1 in 500,000. ■ The genotype and factor activity level in congenital factor VII deficiency do not correlate well with clinical bleeding. ■ Clinical manifestations include epistaxis, mucosal bleeding, soft tissue hematomas, hemarthrosis, menorrhagia, and postoperative bleeding. ■ Thrombosis is a rare complication of congenital factor VII deficiency and is more commonly seen during surgical interventions or replacement therapy.16 8. A. Factor V. Major points of discussion ■ A mixing study with pooled normal plasma should correct the prolonged PT when a factor deficiency is present. ■ Clinical manifestations of congenital factor VII deficiency include epistaxis, mucosal bleeding, soft tissue hematomas, hemarthrosis, menorrhagia, postoperative bleeding, and uncommonly, thrombosis. ■ Recombinant factor VIIa at a low dose of 20 to 30 μg/kg is used to treat congenital factor VII deficiency. ■ Although plasma infusions can be used for treatment in this setting, there is a risk for volume overload, other transfusion reactions, and transfusion transmitted infection. 9. A. Formation of anti–factor VII antibodies. Major points of discussion ■ Although plasma infusions have been used, there is risk for volume overload and other transfusion reactions, as well as transfusion-transmitted infection. ■ The biological half-life of rFVIIa is 2 hours, so it may have to be administered frequently until bleeding ceases. ■ rFVIIa in vivo action is inhibited by tissue factor pathway inhibitor (TFPI). ■ rFVIIa is also used to treat bleeding in patients with hemophilia A or B with inhibitors or patients with acquired factor VIII or IX inhibitors. 10a. A. Consistent with factor VIII half-life. Major points of discussion ■ Anti–factor VIII antibodies can be detected on coagulation studies by performing a mixing study. A mixing study is performed by mixing one volume of patient plasma with one volume of pooled normal plasma. If a factor deficiency is present, the pooled normal plasma will correct the prolonged aPTT. However, the presence of an alloantibody will inhibit the factor VIII in both the patient plasma and the pooled normal plasma. ■ LACs can also inhibit coagulation by interfering with the phospholipid surfaces that coagulation factors require for their activity. However, the inhibitor activity is usually seen upon immediate mixing and with incubation. In addition, LACs are more likely to cause thrombosis, not bleeding. ■ Anti–factor VIII alloantibodies are generally weaker antibodies than LACs, and with incubation, their inhibition will strengthen. Therefore, the immediate mix may correct, and then with incubation for 1 to 2 hours, the aPTT will prolong as the antibody’s binding is enhanced. ■ The strength of an antibody should be determined using a Bethesda assay, which measures the titer of the antibody. The higher the Bethesda units (BUs), the greater the strength of the antibody. A BU reflects the amount of inhibitor that will inactivate half of the factor activity. ■ If there are fewer than 5 BUs, then overwhelming the inhibitor with increasing factor replacement can be attempted (i.e., give a very large dose of factor VIII). However, if there are more than 5 BUs, then alternate concentrates (e.g., porcine factor VIII) or bypassing agents (e.g., factor VIII inhibitor bypassing activity, prothrombin complex concentrates, recombinant factor VIIa) should be used to treat active bleeding. 10b. A. 750 IU. Major points of discussion ■ Calculated dose in units = [% Change in factor activity desired] × [Body weight (kg)] × 0.5 IU/kg. ■ For this patient, assuming factor VIII activity of 0%, a target of 50%, and body weight of 100 kg: Calculated dose = 50% × 100 kg × 0.5 IU/kg = 2500 IU. ■ The target goal for hemarthrosis, gastrointestinal bleeding, and dental extraction is 50%; major surgery and intracranial hemorrhage should have a target goal of 100%. ■ The half-life of factor VIII is 12 hours, so the maintenance dose (50% of loading dose) should be administered approximately every 12 hours. 11a. A. Factor II. Major points of discussion ■ Hemophilia A is caused by a deficiency in factor VIII. ■ Hemophilia A is inherited in an X-linked recessive manner so that males are affected more than females. Females can develop hemophilia A by inheriting two affected alleles, having Turner syndrome (XO), or having skewed lyonization. ■ Factor deficiencies associated with an elevated aPTT but a normal PT include factors VIII, IX, XI, and XII; prekallikrein; and high-molecular-weight kininogen deficiencies. Deficiencies in prekallikrein, factor XII, and high-molecular-weight kininogen are not associated with an increased risk for bleeding. ■ An elevated PTT caused by a factor deficiency can be corrected in vitro by mixing patient plasma with pooled normal plasma (mixing study) if an inhibitor is not present. ■ Hemophilia A is classified into severe (< 1% activity), moderate (1% to 5%), and mild (> 5% to 30%). • Severe hemophilia is associated with spontaneous bleeding. • Moderate hemophilia is associated with bleeding with minor trauma or surgery. • Mild hemophilia usually requires a greater degree of trauma or invasive procedures to cause bleeding. ■ Clinical manifestations include hemarthrosis, deep soft tissue hematomas, hematuria, intracranial hemorrhage, and gastrointestinal bleeding. 11b. A. 75 IU. Major points of discussion ■ Calculated dose in units = % Change in factor activity desired × Body weight (kg) × 1.0 IU/kg. ■ For this patient, assuming factor IX activity of 0%, weight of 30 kg, and desired factor level of 50%: Calculated dose = 50% × 30 kg × 1.0 IU/kg = 1500 IU. ■ Target goal for hemarthrosis is 50%; major surgery has a target of 100%. ■ The half-life of factor IX is 18 to 24 hours, so the maintenance dose (50% of loading dose) should be administered approximately every 24 hours. ■ In addition, factor IX distributes into the extravascular space, so recovery of factor activity after infusion may not reliably be 100%. Therefore, a correction factor to increase the dose to account for the loss of intravascular recovery may be required based on empirical studies for a specific patient. 12. A. 5 hours. Major points of discussion ■ Replacement therapy with factor IX is used in patients with hemophilia B. ■ Targets of 50% are desirable for joint bleeding, dental procedures, hematuria, and gastrointestinal bleeding. ■ Higher targets of 100% are desirable for soft tissue hematomas, intracranial hemorrhage, and surgery. ■ The half-life of factor IX is 18 to 24 hours, which means a loading dose should be provided and then a maintenance dose (usually one half the loading dose) administered every 24 hours. ■ Administration of recombinant factor IX can have 60% to 80% recovery because of the rapid extravascular distribution of the factor after administration. 13. A. Petechiae. Major points of discussion ■ Coagulation factor deficiencies are inherited in an X-linked recessive (factor VIII or factor IX deficiency), autosomal recessive (factor VII or XI deficiency), or autosomal dominant (vWD) manner. ■ Petechiae, persistent bleeding from superficial cuts, and mucosal bleeding such as gum bleeding characterize platelet-type bleeding. ■ Coagulation factor deficiencies are more commonly seen in men because of sex-linked inheritance. ■ It is important to keep in mind that abnormalities of the blood vessels may also lead to bleeding disorders. 14. A. Factor XII. Major points of discussion ■ Vitamin K is a cofactor for γ-carboxylation of glutamic acid residues of the vitamin K–dependent coagulation proteins. ■ The carboxylation of glutamic acid residues results in the chelation of calcium ions and allows the proteins to bind to the phospholipids on the platelet cell membranes to promote the assembly of coagulation factor complexes on the platelet surface. ■ Without sufficient vitamin K, γ-carboxylation does not take place, the proteins are unable to bind to phospholipids, and coagulation proteins cannot interact, making them functionally inactive. ■ Warfarin exerts its effect by inhibiting vitamin K 2,3-epoxide reductase, thereby blocking regeneration of the active form of vitamin K, leading to a lack of γ-carboxylation. 15. A. VKORC1. Major points of discussion ■ VKORC1 and CYP2C9 are two genes with single-nucleotide polymorphisms that predict variable response to warfarin metabolism. ■ CYP2C9 is a P450 enzyme involved in warfarin metabolism. The variant alleles CYP2C9*2 and CYP2C9*3 metabolize warfarin more slowly and may require a lower initial dose. ■ VKORC1 encodes the vitamin K epoxide reductase gene. Variable alleles are associated with variable metabolism of warfarin. Individuals with the group A haplotype produce less VKORC1, and lower warfarin doses are needed in these patients. ■ The CYP2C9 and VKORC1 alleles are found in different frequencies based on ethnic background. For example, the VKORC1 group A haplotype associated with lower warfarin requirements is present in 89% of Asians, but the group B haplotype is seen more in whites and African Americans.22 16. A. Recombinant activated factor VII, 90 μg/kg. Major points of discussion ■ It is more commonly seen in patients with an Ashkenazi Jewish background. ■ Patients may have a range of clinical manifestations from no bleeding tendency to severe bleeding. Factor XI levels do not correlate well with the bleeding risk. ■ The aPTT is prolonged, and the PT is normal. The prolonged aPTT can be corrected by a 1:1 mix with pooled normal plasma. Homozygotes and compound heterozygotes have factor XI levels of less than 15%. ■ Mild bleeding episodes may not have to be treated. However, factor levels above 30% are generally desired before surgery. ■ If treatment is required, a loading dose of 15 to 20 mL/kg of FFP is administered, followed by a 3 to 6 mL/kg dose every 12 hours. Factor XI has a half-life of approximately 50 hours. ■ For severe bleeding, recombinant factor VIIa has been used, including in patients with factor XI inhibitors. These indications are not approved by the FDA for use with recombinant factor VIIa. 17. A. Factor II (prothrombin). Major points of discussion ■ Factor XII deficiency results in an elevated aPTT with a normal PT. However, factor XII deficiency is not associated with bleeding. Rather, factor XII deficiency is thought to have delayed arterial thrombosis. ■ Factor VII deficiency is associated with bleeding that is treated with recombinant factor VIIa or plasma. ■ Factor XIII cross-links fibrin and stabilizes a clot. Deficiency of factor XIII is associated with bleeding. Screening for factor XIII deficiency is performed by measuring clot stability in 5M urea.20,26 18. A. LAC. Major points of discussion ■ Deficiencies in the contact system proteins can result in an elevated aPTT with a normal PT. A mixing study with pooled normal plasma would correct the aPTT (i.e., deficiency, not an inhibitor). ■ Factor XII, prekallikrein, and high-molecular-weight kininogen deficiencies are not associated with bleeding. ■ Complete normalization of prolonged aPTT following prolonged incubation is characteristic of prekallikrein deficiency and is caused by autoactivation of factor XII (activation of factor XII by factor XIIa). ■ Although autoactivation of factor XII has been described with LACs, normalization of the aPTT into the reference range does not occur. In addition, LACs are not associated with correction after mixing with pooled normal plasma.2 19. A. Factor deficiency. Major points of discussion ■ The one-stage coagulation factor assay is an example of a parallel line bioassay. In this type of assay, the results are reliable when the curves of reference and patient plasma are parallel. If there is nonparallelism, the factor activity results should be considered incorrect. ■ A common reason for nonparallelism is the presence of LACs or other nonspecific inhibitors. ■ If three or more patient dilutions are used and they do not agree within a set tolerance limit, the assay is demonstrating an inhibitor pattern. A nonspecific inhibitor should be suspected. ■ The presence of anticoagulants will interfere with clotting times and may underestimate factor activities. ■ The presence of LACs will interfere with coagulation factor activity and can underestimate coagulation factor activity. ■ Elevated factor VIII levels (acute-phase reactant) can shorten the PTT and thus overestimate factor activity. ■ If nonparallelism is encountered using a factor assay with multiple dilutions, a chromogenic factor assay should be performed.25 20. A. Factor II. Major points of discussion ■ Systemic amyloidosis is associated with acquired factor X deficiency. ■ Amyloid fibers are thought to bind directly to factor X. ■ Treatment consists of FFP or prothrombin complex concentrate infusions. ■ Acquired factor X deficiency can result from vitamin K deficiency or liver disease, although the deficiency is usually not isolated; it is usually associated with decreased factors II, VII, and IX. ■ Respiratory infections, thymoma, and malignancies (e.g., renal carcinoma, adrenal adenocarcinoma, acute myeloid leukemia) have also been reported to be associated with acquired factor X deficiency. ■ Congenital factor X deficiency is rare (1 per 500,000). Acquired inhibitors to factor X are uncommon. 21. A. α2-Antiplasmin deficiency. Major points of discussion ■ Factor XIII deficiency is a rare, autosomal recessive disorder. Factor XIII cross-links fibrin through peptide bonds. The clots from these patients dissolve rapidly in 5M urea or in 1% monochloracetic acid. ■ Factor XIII deficiency is associated with delayed umbilical stump bleeding, bleeding after circumcision, hematomas, soft tissue bleeding, and recurrent spontaneous abortions. ■ Factor XIII deficiency is treated with cryoprecipitate or factor XIII concentrates (Europe). ■ Patients with α2-antiplasmin deficiency have a bleeding tendency because of decreased inactivation of plasmin, which leads to excess fibrinolysis. ■ The PT and aPTT are normal in α2-antiplasmin deficiency. Solubility in 5M urea is normal. However, the euglobulin lysis time is shortened. Euglobulin is formed when plasma is acidified, and it contains important fibrinolytic factors such as plasminogen, tPA, and α2-antiplasmin. Deficiency will result in a markedly accelerated lysis time. ■ Fibrinolytic disorders are treated with antifibrinolytics, such as ε-aminocaproic acid.12 22. A. 40 to 120 seconds. Major points of discussion ■ Heparin binds to AT and accelerates its activity 1000-fold, leading to the inactivation of coagulation factors IIa, Xa, IXa, and XIa. ■ UFH is the most common injectable anticoagulant and is used for the treatment of acute coronary syndromes, venous thromboembolism (VTE), and atrial fibrillation. It is also used as the anticoagulant in cardiac bypass and extracorporeal membrane oxygenation circuits. ■ UFH is monitored by following the aPTT. The sensitivity of the aPTT to heparin concentration varies from lot to lot of aPTT reagent. Therefore, the therapeutic range should be established with each new lot of aPTT reagent. ■ The Brill-Edwards method of establishing the UFH therapeutic range requires samples from patients currently on UFH therapy, but not on warfarin (the PT should be in the reference range). UFH anti-Xa activity and the aPTT are measured on each sample. A linear regression is performed; the aPTT corresponding to 0.3 to 0.7 U/mL anti-Xa activity is read from the regression line. The aPTT range is set as the therapeutic range. If heparin-protamine titration is used, then 0.2 to 0.4 U/mL heparin would be used to determine the therapeutic range. ■ UFH is usually administered as a bolus dose and then a constant infusion. The half-life of heparin is 1 to 2 hours. Therefore, heparin therapy is usually monitored by measuring the aPTT every 6 hours while it is adjusted. ■ Adverse reactions to heparin include heparin-induced thrombocytopenia, osteopenia, and hyperkalemia. ■ Heparin is reversed by administered protamine; FFP is not used to reverse heparin.11 23. A. 45-year-old man with a second cousin who had a venous thrombosis. Major points of discussion ■ Acquired risk factors for thrombophilia include malignancy, older age, estrogen replacement, immobilization (e.g., orthopedic surgery), chronic inflammation, and antiphospholipid antibodies. ■ Laboratory testing is recommended only for a select population of patients. ■ Indications for testing for patients with VTE: • Spontaneous thrombosis in a young patient (< 50 years old) • Recurrent VTE • First-degree relatives with VTE • Thrombosis at unusual sites, such as mesenteric vein, portal vein, splenic vein, renal vein, and cerebral sinus, without other risk factors ■ Laboratory testing for thrombophilia can include APCR/factor V Leiden, proteins C and S deficiency, prothrombin G20210A mutation, AT deficiency, homocysteine levels, elevated factor VIII activity, antiphospholipid antibodies (e.g., LAC, aCL antibodies, anti–β2-glycoprotein I antibodies).3 24. A. Decreased factor VIII activity. Major points of discussion ■ Acquired causes of protein S deficiency include vitamin K deficiency, warfarin therapy, liver disease, thrombosis, oral contraceptives, and estrogen replacement therapy. ■ PTT-based protein S assays are performed by diluting patient plasma with protein S–deficient plasma. Fixed amounts of APC and factor Va are added. Prolongation of the PTT is proportional to the amount of protein S in the sample (low protein S → less factor Va inactivated → shorter PTT). ■ PTT-based assays can be affected by LACs, APCR/factor V Leiden, elevated factor VIII activity, direct thrombin inhibitors, and heparin therapy. ■ Protein S circulates as free protein S and as C4 binding protein (C4bp)-bound protein S. C4bp-bound protein S is not active. ■ Total protein S = C4bp − protein S + free protein S.27 25a. A. Factor VIII deficiency. 25b. A. Phospholipid independence of the inhibitor. 25c. A. Anti–glomerular basement membrane (anti-GBM) antibody. 25d. A. A history of three consecutive spontaneous abortions and the presence of aCL antibody are diagnostic for APS. Major points of discussion ■ Thrombosis can be venous, arterial, or microvascular. Microvascular thrombosis may manifest as “catastrophic antiphospholipid syndrome” that typically involves multiorgan failure (lungs, brain, kidneys). ■ According to criteria defining APS, APS is present if at least one of the following clinical and one of the following laboratory criteria are met: ■ Clinical criteria: • Vascular thrombosis: one or more clinical episodes of arterial, venous, or small vessel thrombosis. • Pregnancy morbidity: (a) one or more unexplained deaths of a morphologically normal fetus at or beyond the tenth week of gestation; (b) one or more preterm births of a morphologically normal neonate before the thirty-fourth week of gestation because of eclampsia or severe pre-eclampsia, or recognized features of placental insufficiency; (c) three or more unexplained consecutive spontaneous miscarriages before the tenth week of gestation, with maternal anatomic or hormonal abnormalities and paternal and maternal chromosomal causes excluded. ■ Laboratory criteria: • LAC present in plasma, on two or more occasions at least 12 weeks apart • ACL antibody of immunoglobulin G (IgG) and/or IgM isotype in serum or plasma, present in medium or high titer, on two or more occasions, at least 12 weeks apart • Anti–β2-glycoprotein 1 antibody of IgG and/or IgM isotype in serum or plasma, present on two or more occasions at least 12 weeks apart ■ LAC activity is an in vitro phenomenon defined by prolongation of a phospholipid-dependent coagulation test that is not caused by an inhibitor to a specific coagulation factor. Criteria for the laboratory diagnosis of an LAC include the following: • Prolongation of a phospholipid-dependent clotting test • Demonstration of presence of an inhibitor by mixing tests • Demonstration of the phospholipid dependence of the inhibitor Present on two or more occasions 12 weeks apart ■ Two or more phospholipid-based screening tests should be used to detect LACs. Options for these tests include a low phospholipid concentration PTT, dilute Russell viper venom time, kaolin clotting time, and dilute PT. A confirmatory step using high phospholipid concentration, platelet neutralizing agent, or LAC-insensitive reagent should be performed to demonstrate phospholipid dependence. ■ ELISAs are used to detect aCL and anti–β2-glycoprotein 1 antibodies.3,13 26. A. Tranexamic acid. Major points of discussion ■ A small wire is rotated in a well filled with whole blood (or the well is rotated), and an activator is added. In addition, heparin neutralizers can be added so that testing can be performed during cardiac bypass. As the clot forms, the resistance on the wire or cup increases. This resistance (measured in arbitrary millimeter units) is graphed over time to produce the thromboelastogram. ■ Thromboelastography can show specific defects of platelets, coagulation, and/or fibrinolysis. ■ The time from initiation of the test to the beginning of clot resistance is called the R time and indicates the function of coagulation factors. The rate of clot formation is indicated by the time it takes to reach 20 mm of clot strength. ■ The α angle gives information about fibrin production and clot strength. ■ The maximal amplitude achieved reflects platelet function. ■ Excess fibrinolysis is reflected in the formation of a clot that dissolves faster than normal. ■ The R time is increased in this thromboelastogram, which suggests a coagulation factor deficiency. Therefore, plasma transfusions would be the appropriate therapy.7,15 27. A. Tranexamic acid. Major points of discussion ■ The LY30 measures the percentage decrease in amplitude 30 minutes after maximal amplitude. The LY60 measures the percentage decrease in amplitude 60 minutes after maximal amplitude. ■ The reference range for the LY30 is about 7.5% but is activator dependent (e.g., celite activated or whole blood). ■ The LY30 is a measure of clot lysis (i.e., fibrinolysis). ■ Excess fibrinolysis is reflected in the formation of a clot that dissolves faster than normal. ■ Antifibrinolytic medications include aminocaproic acid, aprotinin, and tranexamic acid. 28. A. Tranexamic acid. Major points of discussion ■ A hypercoagulable state can be measured using TEG by detecting a decreased R time and increased maximal amplitude (MA). ■ Causes of hypercoagulability include the postoperative state, oral contraceptives, pregnancy, deficiencies of natural anticoagulants, and cancer. ■ Thrombocytosis can increase coagulability on TEG by increasing the MA. ■ Hypercoagulability on TEG may suggest a role for antiplatelet agents (e.g., aspirin) or anticoagulants (e.g., heparin, warfarin).7,15 29. A. Chromogenic amidolytic. Major points of discussion ■ There are several assays that measure AT antigen levels. ELISA-based assays, immunodiffusion, and latex particle immunoturbidometric methods are the most common. ■ Type I AT deficiency is a decreased level of functionally normal AT. Antigen assays would be low, and functional assay would be low (i.e., quantitative defect). ■ Type II AT deficiency is a functionally abnormal AT present in normal quantities. Antigen assays would be normal, and functional assay would be low (i.e., qualitative defect). ■ Argatroban, a direct thrombin inhibitor (DTI), will affect thrombin-based functional assays, but not factor Xa–based functional assays.28 30. A. Antigenic-based fibrinogen. Major points of discussion ■ Dysfibrinogenemias can be congenital or acquired. The most common causes of acquired dysfibrinogenemia are associated with liver disease. ■ Among dysfibrinogenemias, 55% are asymptomatic, 25% are associated with bleeding only, and 20% are associated with thrombosis with or without bleeding. ■ The thrombin time is performed by adding a low concentration of thrombin to platelet-poor plasma and measuring the time to clot formation. It is sensitive but is not a very specific test for dysfibrinogenemias. It can be prolonged in the presence of heparin, fibrin degradation products, hypofibrinogenemia, antibovine thrombin antibodies (secondary to fibrin glue), and others. ■ The reptilase time is performed by adding Bothrops atrox snake venom to platelet-poor plasma and measuring the time to clot formation. Reptilase time is prolonged in similar situations such as thrombin time, but it is not sensitive to heparin inhibition. Therefore, a prolonged thrombin time with normal reptilase time suggests the presence of heparin. ■ Antigenic measurement of fibrinogen can be performed with immunoassays. Decreased functional fibrinogen with normal antigenic fibrinogen suggests the presence of a functional abnormality of fibrinogen.10 31. A. 0.5%. Major points of discussion ■ Factor V Leiden (FVL) is an inherited thrombophilia that increases the risk for VTE. ■ The FVL mutation is a mutation of factor V at the APC cleavage site (1691G>A). APC is 10 times slower in inactivating FVL than wild-type factor V. ■ FVL is the most common mutation (> 90%) causing APCR. APCR is caused by other factor V mutations (i.e., FV Cambridge, FV Liverpool, R485K mutation, R2 haplotype, A/G allele). ■ FVL is the most common genetic risk factor for VTE. It is found in 20% to 25% of patients with VTE and 50% of cases of familial VTE. ■ The FVL mutation is seen in 3% to 8% of the general American and European populations. However, the prevalence of the FVL mutation is much lower in African Americans, Native Americans, and Asian Americans.14 32. A. Increased heparin cofactor II. Major points of discussion ■ AT is a natural anticoagulant that inactivates thrombin and factors Xa, IXa, XIa, and XIIa. ■ Administration of UFH can accelerate AT activity 1000-fold. ■ Congenital AT deficiency is seen in 0.02% to 0.17% of the general population and 0.5% to 4.9% of patients with VTE (i.e., very rare). ■ AT levels are decreased in liver disease, active thrombosis, surgery, DIC, sepsis, inflammatory bowel disease, l-asparaginase therapy, nephrotic syndrome, oral contraceptive therapy, and pregnancy. ■ AT levels can be increased by warfarin and argatroban anticoagulation as an artifact of function-based assays.28 33. A. Protein S level of 20%. Major points of discussion ■ First-generation assays were subject to false-negative and false-positive results because of factor deficiencies, the presence of heparin, warfarin therapy, and LACs. ■ Second-generation, modified functional APCR assays are performed after diluting the sample with factor V–deficient plasma to replace any factor deficiencies, thus removing the effect of warfarin therapy or protein C deficiency. The assay also adds polybrene to neutralize unfractionated and low-molecular-weight heparin. ■ Factor V–deficient plasma could have low protein S activity because of the manufacturing process. Therefore, protein S deficiency can still produce false-positive results. ■ LACs are still problematic in second-generation assays, although dilution with factor V–deficient plasma dilutes the antibody and its effect. ■ APCR assays are 100% sensitive for detecting factor V Leiden.29 34. A. Elevated factor VIII activity. Major points of discussion ■ Protein C deficiency can be inherited, acquired, or an artifact of the testing system. ■ Inherited causes of protein C deficiency are seen in 0.14% to 0.5% of the general population and in 3% of patients with a first VTE; it increases the risk for VTE by sevenfold. ■ Acquired causes of protein C deficiency include vitamin K deficiency, warfarin therapy, liver disease, thrombosis, surgery, DIC, and l-asparaginase therapy. ■ In aPTT-based assays, protein C levels may be overestimated when there is an LAC or DTI. aPTT-based assays may also underestimate protein C activity in the presence of increased factor VIII activity and FVL. ■ Chromogenic assays are not subject to the same artifacts as clot-based assays.28 35. A. Double heterozygous protein S and AT deficiency. Major points of discussion ■ Purpura fulminans is characterized by ecchymotic skin lesions that spontaneously become necrotic. Purpura fulminans is often associated with DIC. ■ Protein C is a vitamin K–dependent natural anticoagulant. Activated protein C, with its cofactor protein S, inactivates factors VIIIa and Va. ■ Heterozygous hereditary protein C deficiency is seen in 3% of patients with their first VTE and in only 0.14% to 0.5% of the general population. ■ Protein C deficiency increases the risk for VTE by sevenfold.5 36. A. Activation of osteoclasts. Major points of discussion ■ Osteoporosis caused by osteoclast activation and osteoblast suppression can develop, especially with long-term therapy with low-molecular-weight heparin. ■ Less common complications include increased liver enzymes, alopecia, and hyperkalemia. ■ Administering protamine sulfate reverses heparin anticoagulation.8 37. A. 1.45 Major points of discussion ■ The PT measures extrinsic coagulation factors and is used to monitor warfarin therapy. Warfarin anticoagulation prevents the carboxylation of vitamin K–dependent coagulation factors (factor II, VII, IX, and X and proteins C and S). ■ The sensitivity of each thromboplastin reagent to vitamin K–dependent coagulation factors can differ significantly from manufacturer to manufacturer and lot to lot. The INR was developed so that patients taking warfarin could have their warfarin therapy monitored by different laboratories. ■ Each thromboplastin should have an ISI determined by testing it against an international or working reference preparation, which are thromboplastins available in several different preparations (e.g., rabbit, human, recombinant) from the World Health Organization. ■ To determine the INR of a patient’s PT, the geometric mean of the normal donor population for an individual laboratory must be determined by testing 120 normal donors. The arithmetic mean is not acceptable. ■ INR = (PT/PTGeoMean)ISI. ■ Log (INR) = ISI × PT/ PTGeoMean. ■ PT/ PTGeoMean is often referred to as the PT ratio. ■ The target INR depends on the patient’s condition; in most cases, the treatment goal is an INR between 2 and 3. 38. A. LAC. Major points of discussion ■ The aPTT is usually used to monitor UFH therapy. After a bolus dose and infusion, the aPTT is checked after 6 hours to determine whether the aPTT is in the therapeutic range. Depending on the aPTT, heparin can be adjusted to achieve the target based on being over, under, or at target. ■ However, if heparin is administered and the aPTT does not increase, heparin resistance should be considered. Usually, if more than 25,000 to 35,000 IU of heparin is administered in a 24-hour period without achieving the therapeutic target (as measured by aPTT or activated clotting time) constitutes heparin resistance. Anti-Xa activity of the heparin can be determined. If the anti-Xa is elevated out of proportion to the measured aPTT, then heparin resistance should be considered. ■ Common causes for heparin resistance are elevated factor VIII activity (acute-phase reactant), congenital or acquired AT deficiency, increased heparin clearance, and increased heparin-binding proteins. ■ Treatment can consist of switching to an alternate anticoagulant such as argatroban. In addition, administering AT through infusion of plasma or AT concentrates may be useful. Monitoring with anti-Xa activity instead of aPTT may be useful. 39. A. FFP. Major points of discussion ■ UFH has a half-life of 1 hour, so it takes 5 to 6 hours to be cleared. ■ UFH can be reversed by administering protamine, which is an arginine-rich (positively charged) protein that binds to the negatively charged heparin. ■ Dosing of protamine for heparin reversal: Administer 100 mg of protamine for each 100 IU/mL of heparin activity. ■ The main side effect of protamine is allergic reactions that can be severe (anaphylaxis). It can also cause hypotension, bradycardia, and hypertension/pulmonary hypertension. ■ Protamine is also used to reverse the anticoagulant effect of low-molecular-weight heparin, but it only partially reverses the effect of low-molecular-weight heparin. 40. A. 0.1 Major points of discussion ■ The sensitivity of each thromboplastin reagent to vitamin K–dependent coagulation factors can differ significantly from manufacturer to manufacturer and lot to lot. The INR was developed so that patients taking warfarin could have their warfarin therapy monitored by different laboratories. ■ Each thromboplastin should have an ISI determined by testing it against an international or working reference preparation, which are thromboplastins available in several different preparations (e.g., rabbit, human, recombinant) from the World Health Organization. ■ To determine the ISI of a reagent, the PT is performed using both the international reference preparation or working reference preparation on a group of normal donors and patients on stable warfarin therapy. A double logarithmic plot of a donor/patient’s PT using the reference and test reagent is plotted, and a linear regression is calculated. The ISI is determined in terms of this slope. ■ ISI of test reagent = ISI of the reference preparation × Slope. ■ In this example, ISI = 1.0 × 0.95 = 0.95. ■ The ISI is used to determine the international normalized ratio: • Log (INR) = ISI × PT/ PTGeoMean. • PT/ PTGeoMean is often referred to as the PT ratio.23 41. A. Avascular necrosis. Major points of discussion ■ Warfarin skin necrosis typically appears 3 to 10 days after warfarin initiation if heparin is not overlapped. Lack of the overlap results in clot formation and necrosis of skin, which is more common on the penis, thighs, buttocks, and breasts. It is more common in obese, middle-aged women. ■ Purple toe syndrome is a rare complication appearing 3 to 8 weeks after initiation of therapy. Cholesterol emboli deposit in the skin of the feet, causing pain and the purple color. ■ Most adverse events associated with warfarin are related to hemorrhage, which is seen in 1% to 3% of patients treated with warfarin. There are many drug-drug interactions that may increase the risk for bleeding. Bleeding can be mild with bruising and epistaxis, or it can be severe with intracranial hemorrhage and stroke, gastrointestinal bleeding, intraabdominal bleeding, and compartment syndrome. ■ Osteoporosis and allergic reactions can also occur. 42. A. Pre-anhepatic. Major points of discussion ■ The stages of liver transplantation are: • Pre-anhepatic (surgical dissection and mobilization) • Anhepatic to reperfusion (from occlusion of hepatic vasculature to reperfusion) • Postreperfusion • Postoperative ■ The hemostatic changes associated with each stage of transplantation have been described. ■ A mild deterioration in baseline coagulopathy is noted in the pre-anhepatic stage and is mostly related to the preexisting coagulopathy. ■ The anhepatic to reperfusion stage is associated with a loss of coagulation factor synthesis and clearance and hyperfibrinolysis that can lead to severe bleeding. There is also an increase in tPA that may be caused by reduced clearance and increased release from the ischemic endothelium of the donor liver. ■ During the postreperfusion stage, there is restoration of coagulation factor synthesis and clearance by the transplanted liver, resolution of hyperfibrinolysis, thrombocytopenia, and a heparin-like effect from the donor liver. The resolution of hyperfibrinolysis may be attributed to increased clearance of tPA and increased production of plasminogen activator inhibitor 1. Thrombocytopenia may be caused by sequestration of platelets in the sinusoids of the donor liver. And the heparin-like effect from the donor liver is associated with heparinization of the donor before harvest and the release of heparinoids from the ischemic donor liver endothelium. ■ The postoperative stage is characterized by thrombocytopenia and hypercoagulability. Thrombocytopenia may be caused by platelet activation/consumption in the graft and sequestration owing to hypersplenism. Hypercoagulability is attributed to the early recovery of procoagulants and elevated factor VIII with concomitant delayed recovery of AT, protein C, and protein S. In addition, the hypercoagulability may be exacerbated by the use of antifibrinolytics.24 43. A. Endothelial cell cytotoxicity and disturbance of vascular hemostatic mechanisms. Major points of discussion ■ Heparin is a cofactor that accelerates the binding of AT to thrombin and factor Xa. ■ AT deficiency is associated with an increased risk for VTE. ■ AT deficiency can be inherited or acquired (e.g., liver disease, sepsis, nephrotic syndrome, cardiothoracic surgery/bypass). ■ AT deficiency is associated with heparin resistance. In these cases, therapeutic targets of the activated clotting time or aPTT can be achieved after administering plasma or AT concentrates.6,19 44. A. Abnormal fibrinogen neutralizes tissue plasminogen activator (tPA). Major points of discussion ■ The pattern of inheritance is autosomal dominant. ■ Dysfibrinogenemias classically show an elevated thrombin time and an elevated reptilase time. An elevated thrombin time and normal reptilase time would suggest the presence of heparin. ■ Clinically, approximately half of the patients with dysfibrinogenemias are asymptomatic, and the other half present with bleeding or thrombosis, or both. ■ Two mechanisms used to explain most cases of thrombosis associated with dysfibrinogenemia are (1) abnormal thrombin binding sites on fibrin results in elevated thrombin levels and increased clot formation; and (2) abnormal fibrinogen forms a fibrin clot that is resistant to plasmin degradation.4,6 45. A. Failure to generate APC. Major points of discussion ■ APCR is an inherited thrombophilia caused by an abnormal factor V that is resistant to inactivation by APC. ■ The factor V Leiden (FVL) gene mutation is the most common cause of APCR. Approximately 5% of North American Caucasians carry the FVL gene. ■ In patients with an FVL mutation, APC fails to inactivate factor Va because of a highly conserved point mutation (G1628A), leading to an amino acid substitution (R485K) in factor V, leading to APCR and an increased risk for thrombosis. ■ The factor V Liverpool variant is caused by a point mutation (T1250C), resulting in an amino acid substitution (I359T) that also leads to APCR and an increased risk for thrombosis. ■ The factor V Cambridge variant is caused by a point mutation (G1091C), resulting in an amino acid substitution (R306T) that leads to APCR; however, it is not associated with an increased risk for thrombosis.6 46. A. Failure to inhibit thrombin. Major points of discussion ■ HCII requires heparin, heparan sulfate, or dermatan sulfate as a cofactor. ■ HCII appears to act as an adjunct to AT, and evidence suggests that its deficiency alone does not lead to an increased risk for thrombosis; however, it may increase the risk for thrombosis when there is a concomitant AT deficiency. ■ HCII activity can be measured in vitro by measuring its anti–factor IIa activity in the presence of dermatan sulfate. Alternatively, HCII can be measured antigenically using an ELISA-based method. ■ Testing for HCII deficiency is not recommended as part of a thrombophilia evaluation.6,21 47. A. Protein C. Major points of discussion ■ The liver is the major site of synthesis of both procoagulant and anticoagulant proteins. ■ The procoagulant factors produced by the liver include factors II (prothrombin), VII, IX, X, XI, XII, V, and VIII, plasminogen activator inhibitor 1 (PAI-I), α2-antiplasmin, and thrombin activatable fibrinolysis inhibitor (TAFI). ■ The anticoagulant factors produced by the liver include AT, protein C, protein S, TFPI, and plasminogen. ■ Factor V, factor VIII, PAI-1, AT, protein C, protein S, and TFPI are also synthesized by extrahepatic sites. ■ Hemostatic changes found in liver disease lead to rebalancing of the coagulation system; reduction of procoagulant and fibrinolytic factors is offset by a concomitant decrease of anticoagulant and antifibrinolytic proteins. ■ Patients with liver disease therefore may be at risk for both bleeding and thrombotic complications.24 48. A. Factor VIII. Major points of discussion ■ The liver is the major site of synthesis of both procoagulant and anticoagulant proteins. ■ The procoagulant factors produced by the liver include factors II, VII, IX, X, XI, XII, V, and VIII, PAI-I, α2-antiplasmin, and TAFI. ■ The anticoagulant factors produced by the liver include AT, protein C, protein S, TFPI, and plasminogen. ■ Factor V, factor VIII, PAI-1, AT, protein C, protein S, and TFPI are also synthesized by extrahepatic sites. ■ Hemostatic changes found in liver disease lead to rebalancing of the coagulation system; reduction of procoagulant and fibrinolytic factors is offset by a concomitant decrease of anticoagulant and antifibrinolytic proteins. ■ Patients with liver disease therefore may be at risk for both bleeding and thrombotic complications. 49. A. Failure to generate APC. Major points of discussion ■ Hyperhomocysteinemia is an inherited thrombotic disorder caused by a metabolic defect. ■ Homocysteine is an intermediate amino acid formed during the conversion of methionine to cysteine; the enzyme cystathionine-β-synthase is required. ■ Moderate elevations of homocysteine may be a risk factor for atherothrombotic disease and VTE. ■ Elevation of homocysteine can be caused by inherited defects of metabolism, nutritional deficiencies of vitamin cofactors, acquired diseases, and medications. ■ Vitamin supplementation with folate, vitamin B6, and vitamin B12 can decrease homocysteine levels, but their clinical usefulness as prophylaxis against arterial and venous thrombotic events is questionable.6 50. A. There is good evidence to support the use of routine coagulation tests for predicting bleeding or thrombotic risk in liver disease. Major points of discussion ■ The liver is the major site of synthesis of both procoagulant and anticoagulant proteins. ■ The procoagulant factors produced by the liver include factors II, VII, IX, X, XI, XII, V, and VIII, PAI-I, α2-antiplasmin, and TAFI. ■ The anticoagulant factors produced by the liver include AT, protein C, protein S, TFPI, and plasminogen. ■ Factor V, factor VIII, PAI-1, AT, protein C, protein S,and TFPI are also synthesized by extrahepatic sites. ■ Hemostatic changes found in liver disease lead to rebalancing of the coagulation system; reduction of procoagulant and fibrinolytic factors is offset by a concomitant decrease of anticoagulant and antifibrinolytic proteins. ■ Patients with liver disease therefore may be at risk for both bleeding and thrombotic complications. ■ Although there is currently no evidence that routine coagulation tests are useful for predicting bleeding or thrombotic risk in liver disease, investigators are exploring the value of global coagulation assays such as thrombin generation and thromboelastography in this clinical scenario.24 51. A. Protein C deficiency. Major points of discussion ■ tPA activates plasminogen to plasmin. Plasmin degrades fibrinogen and fibrin to its degradation products. ■ Excess PAI-1 activity decreases tPA levels and decreases plasminogen activation, leading to decreased plasmin and decreased fibrinolysis. ■ Excess PAI-1 activity can lead to arterial and venous thrombotic disorders, such as VTE and myocardial infarction. ■ Congenital deficiencies of PAI-1 lead to a bleeding disorder because tPA is inadequately inactivated and leads to excess fibrinolysis.6 52. A. Failure of APC to inactivate factors Va and VIIIa. Major points of discussion ■ Plasminogen is a zymogen that is activated to plasmin by the serine proteases tPA and urokinase. ■ Plasmin degrades fibrinogen and fibrin with the primary role of dissolving thrombi. ■ Both homozygous and heterozygous forms of plasminogen deficiency have been described; they can be qualitative defects (active site mutations; type II) or quantitative defects (type I). It is a very rare disorder. ■ Patients can be asymptomatic or present with a history of thrombosis. They can also have ligneous conjunctivitis with pseudomembranes covering the eyes (other organs can also be affected). The pseudomembranes are caused by the accumulation of fibrin and are more commonly found in patients with type I deficiencies. ■ It has not been clearly established if plasminogen deficiency alone is sufficient to cause a hypercoagulable state.6,17 53. A. Failure to inhibit factor IIa, factor Xa, and other activated factors. Major points of discussion ■ Thrombomodulin on endothelial surfaces accelerates the activation of protein C to APC. ■ APC, along with its cofactor protein S, inactivates factors Va and VIIIa. ■ Protein C deficiency is a rare inherited disorder that is associated with an increased risk for VTE. Type I defects are quantitative defects of normal protein C and type II defects are qualitative defects. ■ Protein C deficiency should not be confused with APCR, which is caused by a mutation in factor V that makes it resistant to inactivation by APC. The most common mutation causing APCR is FVL, which may be present in up to 50% of North American Caucasians.6 54. A. Failure to inhibit thrombin. Major points of discussion ■ Protein S circulates bound to C4 binding protein (C4bp) or as free protein S (Total protein S = Free protein S + Protein S:C4bp). ■ Approximately 65% of protein S is bound to C4bp, and the remainder is free protein S. Only the free form has cofactor activity. ■ Protein S deficiency is an inherited, autosomal dominant disorder that is a risk factor for VTE. However, inherited protein S deficiency is not common. ■ Assays for protein S include total protein S antigen, free protein S antigen, and total protein S activity. Antigenic assays are immunologically based, whereas activity is measured by clot-based assays using platelet poor plasma. ■ Type I protein S deficiency is a quantitative disorder of normal protein S. Type II is a qualitative disorder of quantitatively normal protein S. Type III is a disorder of normal total protein S, but decreased free protein S and increased bound protein S. ■ It is important to note that there is a physiologic decrease in protein S during pregnancy. Therefore, a physiologic decrease in protein S should not be misinterpreted as protein S deficiency. Other possible causes of decreased protein S activity include warfarin therapy, vitamin K deficiency, and liver disease.6,27 55. A. Endothelial cell toxicity and disruption of vascular hemostatic mechanisms. Major points of discussion ■ tPA converts plasminogen to plasmin. Plasmin functions to degrade fibrinogen and fibrin. ■ Recombinant tPA is a pharmaceutical agent that is used therapeutically for acute ischemic cerebrovascular events, pulmonary embolism, and other thrombotic disorders. ■ Increased endogenous tPA activity can lead to excessive fibrinolysis and bleeding. ■ tPA deficiency can lead to hypofibrinolysis and a risk for thrombosis.
Coagulation
Hemostasis and Thrombosis (Anticoagulation, Thrombophilias, Fibrinolysis)
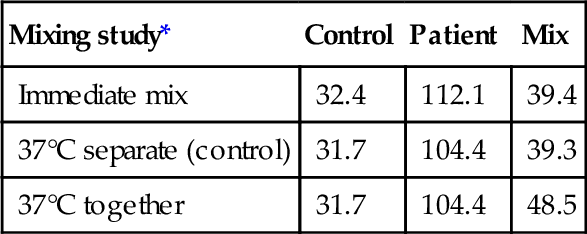
Mixing study*
Control
Patient
Mix
Immediate mix
32.4
112.1
39.4
37°C separate (control)
31.7
104.4
39.3
37°C together
31.7
104.4
48.5
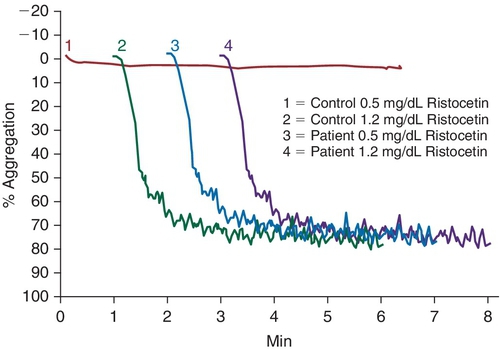
Rationale: Similar levels in young children and adults.
B. Factor II.
C. Factor X.
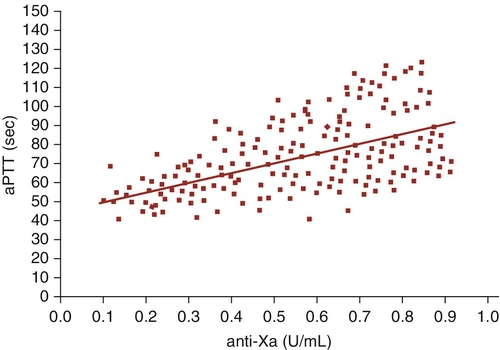
D. Factor XI.
E. Prekallikrein.
Rationale: Lower levels in young children than adults.
Rationale: The DDAVP challenge is a clinical test to measure increased vWF:Ag and vWF:RCo activity after the administration of DDAVP. DDAVP is contraindicated in platelet-type vWD.
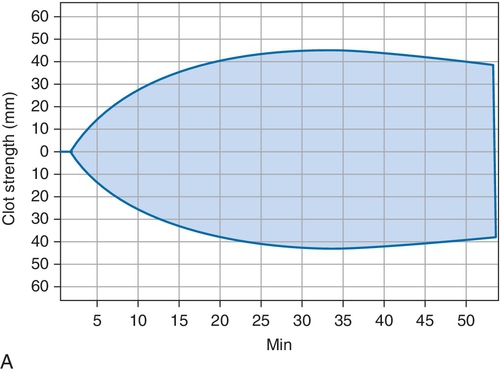
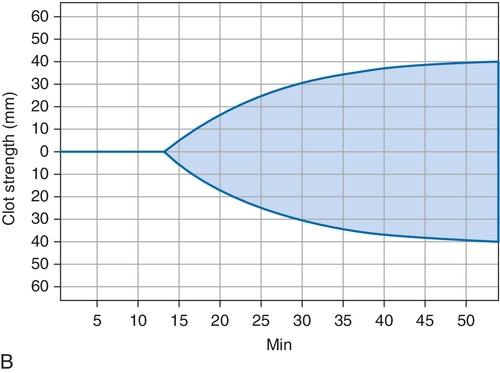
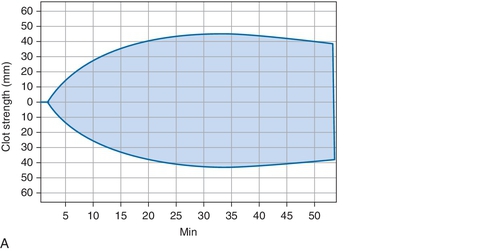
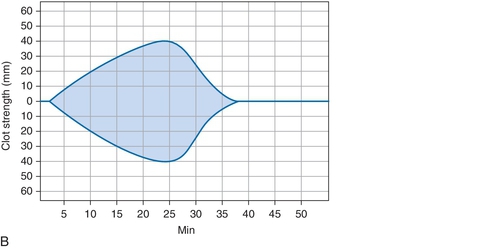
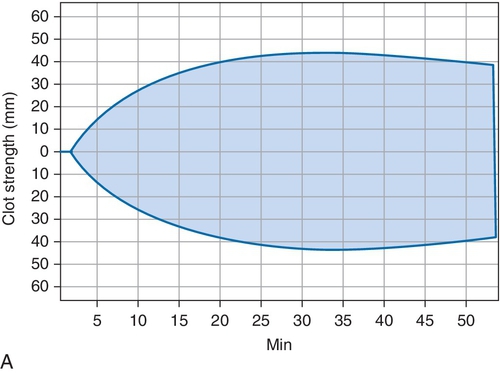
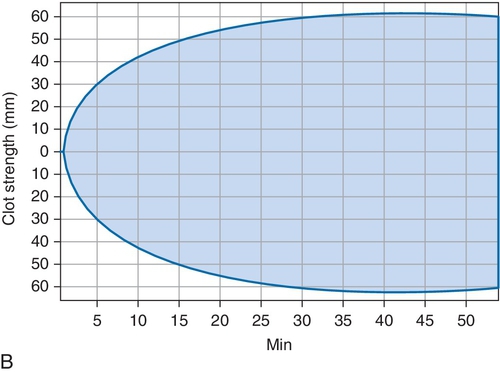
B. Cryoprecipitate challenge.
Rationale: Platelet-type vWD (PT-vWD) and type 2B vWD can present with similar clinical and laboratory findings. PT-vWD is caused by gain-of-function mutation in the platelet glycoprotein GPIbα that leads to increased binding to vWF. Type 2B vWD is caused by gain-of-function mutation in the platelet-binding domain of vWD. Although presentation can be variable, both disorders can have thrombocytopenia, increased bleeding time, decreased FVIII activity, and increased vWF:Ag/RCo ratio. Both also aggregate with low concentrations of ristocetin, whereas normal controls do not. However, the addition of cryoprecipitate containing normal vWF leads to spontaneous aggregation of PT-vWD platelets, whereas type 2B vWD will not.
C. Platelet function analyzer (PFA)-100.
Rationale: This is a screening test that can detect vWD, but it cannot distinguish between type 2B vWD and PT-vWD.
D. Partial thromboplastin time (PTT) mixing study.
Rationale: If the PTT is elevated because of decreased FVIII activity, then the mixing study will correct. Both type 2B and PT-vWD can present with decreased FVIII activity.
E. von Willebrand factor (vWF) multimer analysis.
Rationale: Type 2B vWD and PT-vWD have loss of high-molecular-weight vWF multimers.
Rationale: Results consistent with type 3 vWD are markedly decreased vWF antigen level; markedly decreased vWF activity; markedly decreased factor VIII activity; multimer analysis demonstrating an absence of high, intermediate, and small bands; and absent ristocetin-induced platelet aggregation.
B. Type 2N.
Rationale: Results consistent with type 2N vWD are normal vWF antigen level; normal vWF activity; markedly decreased factor VIII activity; multimer analysis demonstrating high, intermediate, and small bands; and normal ristocetin-induced platelet aggregation.
C. Type 2A.
Rationale: Results consistent with type 2A vWD are normal to mildly decreased vWF antigen level, vWF activity lower than the antigen level, normal factor VIII level, multimer analysis demonstrating absent high and intermediate bands, and decreased ristocetin-induced platelet aggregation.
D. Type 2M.
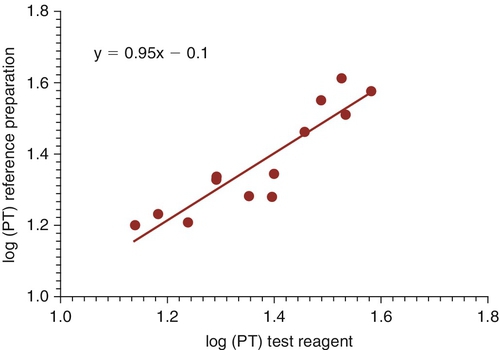
Rationale: Results consistent with type 2M vWD are normal to mildly decreased vWF antigen level; vWF activity lower than the antigen level; normal factor VIII level; multimer analysis demonstrating high, intermediate, and small bands; and decreased ristocetin-induced platelet aggregation.
E. Type 1.
Rationale: Results consistent with type 1 vWD are mildly decreased vWF antigen level; mildly decreased vWF activity; mildly decreased factor VIII activity; multimer analysis demonstrating high, intermediate, and small bands; and normal or decreased ristocetin-induced platelet aggregation.
Rationale: Incorrect because blood group O individuals tend to have lower baseline vWF antigen levels than those with other blood groups.
B. At the time of initial evaluation the patient was taking oral contraceptives, which are known to increase vWF levels.
Rationale: vWF is an acute-phase reactant that increases during pregnancy and when taking oral contraceptives.
C. The blood sample for testing was transported to the laboratory at room temperature instead of the preferred temperature of 4°C.
Rationale: Transporting the sample at 4°C would have caused the vWF levels to have been artifactually low because of cryoprecipitation of the vWF. Samples should be transported to the laboratory at room temperature.
D. At the time of the initial evaluation, the patient had not recently exercised, and a lack of physical activity is known to increase vWF levels.
Rationale: Exercise and physical stress are known to increase vWF levels.
E. vWF levels are known to decrease with increasing age.
Rationale: The age-related decrease in vWF levels occurs from birth until approximately 180 days of life. After this time, the levels are the same as in adults.
Rationale: Left ventricular assist devices can cause acquired vWD.
B. Inflammatory bowel disease.
Rationale: Lack of absorption of vitamin K leads to vitamin K deficiency with decreased activity of factors II, VII, IX, and X and proteins C and S; these are the vitamin K–dependent coagulation factors.
C. Phytonadione therapy.
Rationale: Phytonadione is vitamin K, and its administration should increase levels of vitamin K–dependent coagulation factors (factors II, VII, IX, and X and proteins C and S).
D. Heparin therapy.
Rationale: Heparin therapy does not affect levels of factor VII. Heparin therapy can decrease levels of AT.
E. Ganciclovir therapy.
Rationale: Antibiotics decrease gut flora and lead to vitamin K deficiency with decreased levels of factor II, VII, IX, and X; these are the vitamin K–dependent coagulation factors. Ganciclovir is an antiviral and should not lead to factor VII deficiency.
Rationale: Factor VII has a half-life of approximately 5 hours.
B. Factor X.
Rationale: Factor X has a half-life of approximately 40 hours.
C. Factor II.
Rationale: Factor II has a half-life of approximately 65 hours.
D. Factor XIII.
Rationale: Factor XIII has a half-life of approximately 200 hours.
E. Factor VIII.
Rationale: Factor VIII has a half-life of approximately 10 hours.
Rationale: This is a common complication.
B. Thrombosis.
Rationale: This is a rare complication of congenital factor VII deficiency and is associated with surgery and sometimes recombinant factor VIIa therapy.
C. Menorrhagia.
Rationale: Up to 60% of women with congenital factor VII deficiency have menorrhagia.
D. Central nervous system bleeding.
Rationale: Occurs most commonly in the first 6 months of life.
E. Postoperative bleeding.
Rationale: Up to 30% of patient will bleed after surgery.
Rationale: PT and aPTT are generally prolonged in this setting.
B. Factor VII.
Rationale: A normal aPTT and a prolonged PT that corrects with mixing with pooled normal plasma are characteristic of congenital factor VII deficiency.
C. Factor VIII.
D. Factor IX.
E. Factor XI.
Rationale: Hemophilia A, B, and C are characterized by a prolonged aPTT and a normal PT
Rationale: Formation is possible but extremely rare in patients with congenital factor VII deficiency.
B. Short half-life of recombinant factor VIIa.
Rationale: Recombinant factor VIIa must be administered every 2 hours because of the short half-life.
C. Requirement for high doses (120 μg/kg).
Rationale: Low doses (20 to 30 μg/kg) are usually given; this is a much lower dose than for patients with factor VIII inhibitors.
D. Contamination with activated factors II, IX, and X.
Rationale: Activated prothrombin concentrates can have activated factors II, VII, IX, and X. However, recombinant factor VIIa does not contain the activated factors II, IX, and X.
E. Need to overlap with heparin therapy.
Rationale: Initiation of warfarin requires heparin overlap, but recombinant factor VIIa is not used for anticoagulation and does not require heparin overlap.
Rationale: The half-life of factor VIII is 12 hours, so approximately 25% of factor VIII activity should remain after 12 hours (the administered dose should result in 50% peak factor VIII levels). The mixing study in this case is consistent with an inhibitor.
B. Consistent with inadequate dosing of factor VIII.
Rationale: The dose is calculated correctly for factor VIII replacement: body weight (kg) × desired % increase × 0.5 IU/kg.
C. Inadequate reconstitution of lyophilized product.
Rationale: The mixing study is not consistent with a factor deficiency.
D. Presence of a lupus anticoagulant (LAC).
Rationale: An LAC could produce an isolated elevated PTT without correction on a mixing study. However, the time dependence of the antibody and bleeding, rather than thrombosis, suggests that this is less likely an LAC.
E. Factor VIII inhibitor.
Rationale: The percentage recovery of factor activity is too low after 12 hours, and the mixing study shows a time-dependent inhibitor. These findings are most consistent with a factor inhibitor.
B. 1000 IU.
Rationale: These doses are too low for weight and target goal.
C. 2500 IU.
Rationale: This dose may be appropriate for factor VIII replacement.
D. 3500 IU.
E. 5000 IU.
Rationale: These doses are too high. The target should be 50%.
Rationale: Factor II deficiency should affect both the PT and the aPTT.
B. Factor VII.
Rationale: Factor VII deficiency is usually associated with an elevated PT and a normal aPTT.
C. Factor VIII.
Rationale: An isolated aPTT in the setting of clinical bleeding can be caused by factor VIII, factor IX, and factor XI deficiencies.
D. Factor X.
Rationale: The PT and aPTT are usually affected in factor X deficiency.
E. Factor V.
Rationale: The PT and aPTT are usually affected in factor V deficiency.
B. 210 IU.
C. 750 IU.
Rationale: These doses are too low for weight and target goal.
D. 1500 IU.
Rationale: This dose may be appropriate.
E. 5000 IU.
Rationale: This dose is too high.
Rationale: The half-life of factor VII is approximately 5 hours.
B. 12 hours.
Rationale: The half-life of factor VIII is 12 hours.
C. 24 hours.
Rationale: The half-life of factor IX is 18 to 24 hours.
D. 48 hours.
Rationale: The half-life of factor XI is approximately 50 hours.
E. 96 hours.
Rationale: The half-life of fibrinogen is approximately 4 days.
B. Superficial hematomas.
Rationale: Deep hematomas are characteristic of coagulation deficiencies.
C. Epistaxis.
D. Delayed bleeding.
Rationale: Delayed postoperative bleeding is characteristic of coagulation deficiencies.
E. Gum bleeding.
Rationale for A, C, and E: Seen in platelet-type bleeding.
B. Factor II.
Rationale: Factors II, VII, IX, and X and proteins C and S are vitamin K–dependent factors.
C. Factor VIII.
D. Factor V.
E. Factor XIII.
Rationale for A, C, D, and E: These factors are not vitamin K dependent.
Rationale: VKORC1 single-nucleotide polymorphisms are associated with variable metabolism of warfarin.
B. CYP2D9.
Rationale: CYP2C9, not CYP2D9, is a hepatic P450 enzyme associated with variable metabolism of warfarin.
C. ABCB1.
Rationale: Encodes P-glycoprotein, a drug efflux pump, that may be involved in dabigatran availability.
D. CYP3A4.
E. CYP2J2.
Rationale: Involved in the metabolism of rivaroxaban.
Rationale: This may be considered for life-threatening bleeding, but it is not the standard therapy for factor XI deficiency,
B. Fresh frozen plasma (FFP), 15 mL/kg.
Rationale: This is the correct loading dose of plasma for factor XI deficiency.
C. Recombinant factor VIII.
Rationale: The patient most likely has factor XI deficiency.
D. Fibrin glue.
Rationale: Fibrin glue can be used for dental extraction, but not for uterine bleeding.
E. Recombinant factor XI.
Rationale: This is not available as a U.S. Food and Drug Administration (FDA)-approved drug.
Rationale: Factor II deficiency is associated with hematomas and hemarthroses.
B. Factor VII.
Rationale: Congenital and acquired factor VII deficiencies are associated with bleeding.
C. Factor XI.
Rationale: Factor XI deficiency is associated with variable degrees of bleeding from asymptomatic to severe. Factor levels do not predict bleeding manifestations.
D. Factor XII.
Rationale: Factor XII deficiency is not associated with bleeding.
E. Factor XIII.
Rationale: Factor XIII deficiency is associated with bleeding.
Rationale: In the presence of an LAC, the mixing study would not correct and autoactivation would not be seen.
B. Protein C deficiency.
Rationale: Protein C deficiency is not associated with an elevated aPTT, correction of a mixing study, or autoactivation.
C. Factor XIII deficiency.
Rationale: Factor XIII deficiency does not show an elevated aPTT, correction of a mixing study, or autoactivation.
D. Prekallikrein deficiency.
Rationale: Prekallikrein deficiency is associated with increased aPTT (intrinsic pathway), correction with a mixing study (deficiency), and autoactivation.
E. High-molecular-weight kininogen deficiency.
Rationale: High-molecular-weight kininogen deficiency is associated with increased aPTT (intrinsic pathway) and correction with a mixing study (deficiency), but autoactivation is not a characteristic.
Rationale: Agreement among dilutions of the patient sample does not indicate a factor deficiency.
B. Inhibitor pattern.
Rationale: An inhibitor pattern shows nonparallelism. In nonparallelism, dilutions of patient plasma do not provide the same factor activity within allowable error.
C. Noncalibrated curve.
D. Linear range.
Rationale: A calibration curve is determined using dilutions of calibrator, not a patient sample. The linear range is determined after the best-fit curve is fit to the calibration points and the widest linear range.
E. Between-subject variability.
Rationale: The variability in this experiment is within subjects at three patient plasma dilutions. Between-subject variability would be determined between two subjects.
B. Factor VII.
C. Factor VIII.
D. Factor X.
Rationale: This deficiency is associated with systemic amyloidosis.
E. Factor XII.
Rationale for A, B, C, and E: These deficiencies are not associated with systemic amyloidosis.
Rationale: α2-Antiplasmin deficiency would show normal clot dissolution in the 5M urea test.
B. Dysfibrinogenemia.
Rationale: Dysfibrinogenemia would show a prolonged thrombin time.
C. Factor XIII deficiency.
Rationale: The rapid clot dissolution in 5M urea is characteristic of factor XIII deficiency.
D. Fibrinolysis.
Rationale: An elevated d-dimer would be expected if bleeding were caused by fibrinolysis.
E. High-molecular-weight kininogen deficiency.
Rationale: High-molecular-weight kininogen deficiency would cause a prolongation of the aPTT and is not a cause of bleeding.
Rationale: This corresponds to 0.1 to 0.9 U/mL anti-Xa.
B. 40 to 60 seconds.
Rationale: This corresponds to 0.1 to 0.3 U/mL anti-Xa.
C. 60 to 80 seconds.
Rationale: This corresponds to 0.3 to 0.7 U/mL anti-Xa.
D. 60 to 120 seconds.
Rationale: This corresponds to 0.3 to 0.9 U/mL anti-Xa.
E. 80 to 120 seconds.
Rationale: This corresponds to 0.7 to 0.9 U/mL anti-Xa.
Rationale: Testing should be performed when there are first-degree relatives with VTE.
B. 49-year-old man with hypertension, hyperlipidemia, and diabetes with first stroke.
Rationale: Testing should be performed when there is spontaneous thrombosis, in the absence of such risk factors as mentioned previously, in a patient younger than 50 years.
C. 55-year-old man with first mesenteric vein thrombosis.
Rationale: Thrombosis at unusual sites, such as mesenteric vein, portal vein, splenic vein, renal vein, and cerebral sinus, without other risk factors is an indication for thrombophilia testing.
D. 35-year-old woman with first spontaneous abortion in first trimester.
Rationale: Testing should be performed for recurrent spontaneous abortions and abortions in the second and third trimesters of pregnancy.
E. 45-year-old woman taking oral contraceptives.
Rationale: Testing should be performed for women intending to conceive or to start oral contraceptives when there is a first-degree relative with a thrombophilia.
Rationale: Elevated factor VIII levels can falsely decrease protein S activity in PTT-based assays.
B. Decreased factor V activity.
Rationale: Factor V is normalized in the assay with protein S–deficient plasma, and factor Va is also added.
C. Direct thrombin inhibitor.
Rationale: Direct thrombin inhibitors overestimate the protein S level.
D. Factor V Leiden mutation.
Rationale: Factor V Leiden mutation and other causes of APCR can lead to a falsely low protein S activity on a PTT-based protein S activity assay.
E. Pregnancy.
Rationale: Protein S levels decrease with pregnancy, which is a normal physiologic response. It is a true decrease in the protein S activity.
Rationale: aPTT for the 1:1 mix should have corrected into the reference range.
B. Severe factor VII deficiency.
Rationale: There should be a prolongation of the PT.
C. LAC.
Rationale: The prolonged aPTT, failure of the aPTT to correct in the mixing study, and the DVT are consistent with the presence of an LAC.
D. Disseminated intravascular coagulation (DIC).
Rationale: Both the PT and aPTT would be prolonged and thrombocytopenia would be expected in DIC.
E. Factor IX inhibitor.
Rationale: An inhibitor to factor IX would be expected to present with bleeding, not clotting.
Rationale: LACs are phospholipid dependent.
B. Phospholipid dependence of the inhibitor.
Rationale: LACs prolong clotting times by binding to phospholipids and interfering with their ability to provide a surface for the clotting cascade.
C. Mixing study in which the aPTT corrects into the reference range.
Rationale: Because an LAC is an inhibitor, the aPTT should not correct into the reference range.
D. Prolongation of a phospholipid-independent clotting test.
Rationale: A phospholipid-dependent clotting test should be prolonged because LACs interfere with phospholipids.
E. Mixing study where the aPTT prolongation is time dependent.
Rationale: Time-dependent prolongation of the aPTT upon incubation is suggestive of the presence of an inhibitor to a specific coagulation factor, not an LAC.
Rationale: Antibody against type IV collagen found in the GBM and alveolar basement membrane.
B. Antinuclear antibody.
Rationale: Antibody associated with systemic lupus erythematosus (SLE).
C. Anti–Jo-1 antibody.
Rationale: Antibody associated with polymyositis.
D. Anti–ADAMTS-13 antibodies.
Rationale: Antibody associated with idiopathic thrombotic thrombocytopenic purpura.
E. Anti–β2-glycoprotein 1 antibodies.
Rationale: One of three major types of antiphospholipid antibodies.
B. A DVT and positive mixing study showing the presence of an inhibitor are diagnostic for APS.
C. An arterial thrombosis is necessary to diagnose APS.
Rationale: Arterial, venous, and small vessel thromboses are all a part of the clinical criteria for diagnosing APS.
D. An antiphospholipid antibody must be demonstrated on more than one occasion to meet the diagnostic criteria for APS.
Rationale for A, B, and D: As part of the criteria, antiphospholipid antibodies should be detected on two or more occasions, at least 12 weeks apart.
E. Detection of any antiphospholipid antibody is itself diagnostic of APS.
Rationale: Diagnosis of APS requires at least one clinical criterion and one laboratory criterion to be met.
Rationale: Tranexamic acid is an antifibrinolytic agent. The thromboelastogram shows a coagulation deficiency.
B. Aspirin.
Rationale: Aspirin is an antiplatelet agent. The thromboelastogram shows a coagulation deficiency.
C. Packed red blood cell transfusion.
Rationale: Packed red blood cells are used to treat anemia, not hemostatic defects. The thromboelastogram shows a coagulation deficiency.
D. Plasma transfusion.
Rationale: Plasma is used to replace coagulation factors and natural anticoagulants. The thromboelastogram shows a coagulation deficiency, so this would be the appropriate therapy. The R value is prolonged in this case.
E. Platelet transfusion.
Rationale: Platelet transfusions are used to treat quantitative or qualitative platelet disorders. The thromboelastogram shows a coagulation deficiency.
Rationale: Tranexamic acid is an antifibrinolytic agent. This thromboelastogram shows fibrinolysis.
B. Heparin.
Rationale: Heparin would be the most appropriate therapy for a hypercoagulable state. This thromboelastogram shows fibrinolysis.
C. Packed red blood cell transfusion.
Rationale: Packed red cells are used to treat anemia, not hemostatic defects. This thromboelastogram shows fibrinolysis.
D. Plasma transfusion.
Rationale: Plasma is used to replace coagulation factors and natural anticoagulants. This thromboelastogram shows fibrinolysis.
E. Platelet transfusion.
Rationale: Platelets are used to treat quantitative or qualitative platelet disorders. This thromboelastogram shows fibrinolysis.
Rationale: Tranexamic acid is an antifibrinolytic agent. This thromboelastogram shows a hypercoagulable state.
B. Heparin.
Rationale: Heparin would be the most appropriate therapy for a hypercoagulable state.
C. Packed red blood cell transfusion.
Rationale: Packed red cells are used to treat anemia, not hemostatic defects. This thromboelastogram shows a hypercoagulable state.
D. Plasma transfusion.
Rationale: Plasma is used to replace a balance of normal levels of coagulation factors and natural anticoagulants. This thromboelastogram shows a hypercoagulable state, and the levels of natural anticoagulants in plasma would not be therapeutically sufficient.
E. Platelet transfusion.
Rationale: Platelet transfusions are used to treat quantitative or qualitative platelet disorders. This thromboelastogram shows a hypercoagulable state.
Rationale: This is a functional assay.
B. Enzyme-linked immunoassay (ELISA).
C. Latex particle immunoturbidometric.
Rationale: These are measures of antigen, not function.
D. DNA analysis.
Rationale: This is a molecular method that may allow prediction of the phenotype based on the genotype, but does not directly measure activity.
E. Heparin-binding site inhibition.
Rationale: There is no such assay that is routinely available clinically. If there were an existing assay to measure this, it would miss the other types of mutations, such as those of the reactive site.
Rationale: Measures all fibrinogen regardless of functional activity.
B. Fibrometer-based fibrinogen.
Rationale: Can be falsely low in mechanical-based clotting assay for fibrinogen.
C. Nephelometry.
Rationale: A method for antigenic measurement of fibrinogen, which measures all fibrinogen regardless of functional activity.
D. Reptilase time.
Rationale: Not as sensitive as the thrombin time.
E. Thrombin time.
Rationale: Prolonged in all congenital forms of dysfibrinogenemia except fibrinogens Oslo I and Valhalla.
Rationale: 0.45% of Asian Americans have the factor V Leiden mutation.
B. 1%.
Rationale: 1.2% of African Americans and 1.25% of Native Americans have the factor V Leiden mutation.
C. 5%.
Rationale: 5.2% of Caucasian Americans have the factor V Leiden mutation.
D. 15%.
Rationale: In some areas of Europe, the prevalence of the factor V Leiden mutation can be as high as 10% to 15% (e.g., Greece).
E. 25%.
Rationale: This is a very high prevalence; it has not been described in general populations of the United States or Europe.
Rationale: Increases AT activity.
B. l-Asparaginase therapy.
Rationale: Decreases AT activity.
C. Hematuria.
Rationale: Proteinuria, not hematuria, decreases AT levels.
D. Menopause.
Rationale: Postmenopausal levels of AT activity increase.
E. Warfarin therapy.
Rationale: Anticoagulation increases AT activity.
Rationale: Protein S level of less than 20% could cause false-positive results in the first-generation assay, but usually not in the second-generation assay. The second-generation assays normalize vitamin K–dependent factors by the addition of factor V–deficient plasma. However, protein S activity may be low in factor V–deficient plasmas, and this could influence the result.
B. Warfarin therapy.
C. Heparin 0.7 U/mL.
Rationale: Second-generation APCR assays include the addition of polybrene to neutralize unfractionated and low-molecular-weight heparin.
D. LAC.
Rationale: This is the most likely cause of a false-positive result in the second-generation APCR assay.
E. Liver dysfunction.
Rationale for B and E: Second-generation APCR assays normalize vitamin K–dependent factors by the addition of factor V–deficient plasma.
Rationale: Increased factor VIII activity will underestimate the protein C activity.
B. Factor V Leiden.
Rationale: APCR, caused byFVL, will underestimate the protein C activity. It would not affect a dilute Russell viper venom–based assay.
C. LAC.
Rationale: Artifactual elevation of protein C activity can be seen in aPTT-based assays in the presence of LACs.
D. Warfarin therapy.
Rationale: Warfarin inhibits the carboxylation of vitamin K–dependent factors such as protein C so that its activity is reduced.
E. Thrombosis.
Rationale: Thrombosis can incorporate protein C into natural anticoagulant complexes, decreasing the detected protein C activity levels. Testing should not be performed immediately after an acute thrombotic event.
B. Dysfibrinogenemia.
C. Homozygous factor V Leiden.
D. Homozygous protein C deficiency.
Rationale: Severe protein C deficiency as seen in homozygous protein C mutations can present in the newborn period with purpura fulminans and DIC.
E. Homozygous prothrombin 20210G>A mutation.
Rationale for A, B, C, and E: These increase the risk for thrombosis but are not characteristic causes of this presentation.
Rationale: Osteoporosis due to suppression of osteoblasts and activation of osteoclasts develops with unfractionated and, more often, low-molecular-weight heparin.
B. Development of anti–heparin polyvinyl sulfate antibodies.
Rationale: Heparin-induced thrombocytopenia can be a serious complication of heparin therapy. Antibodies against heparin–platelet factor 4 can lead to immune thrombocytopenia. Polyvinyl sulfate complexes with heparin are used in ELISA-based anti–heparin platelet factor 4 assays.
C. Development of Hollenhorst plaques.
Rationale: Cholesterol emboli in the eyes (Hollenhorst plaques) are not a complication of heparin therapy.
D. Hypokalemia.
Rationale: Hyperkalemia is an uncommon side effect of heparin therapy.
E. Hirsutism.
Rationale: Alopecia, not hirsutism, can be seen with heparin therapy.
Rationale: This is the ratio of PT to arithmetic mean. It is not the INR.
B. 1.47
Rationale: This is the ratio of the PT to geometric mean. It is not the INR.
C. 1.59
Rationale: INR = (PT/geometric mean)ISI.
D. 1.62
Rationale: This is (PT/arithmetic mean)ISI. It is not the INR.
E. 5.67
Rationale: This is the PT/standard error. It is not the INR.
Rationale: Although an LAC increases the risk for DVT, it does not explain the heparin resistance. LACs usually prolong the PT and/or aPTT.
B. AT deficiency.
Rationale: Inherited or acquired AT deficiency affects heparin therapy because heparin is not able to accelerate AT effectively if AT is present in low amounts.
C. Acquired factor VIII deficiency.
Rationale: The aPTT should be prolonged before initiation of therapy. Heparin resistance is seen with elevated factor VIII activity.
D. APCR.
Rationale: APCR, most commonly caused by the FVL mutation, is a risk factor for DVT. However, it should not lead to heparin resistance.
E. Increased heparin cofactor II activity.
Rationale: Heparin cofactor II binds heparin, which leads to inactivation of factor IIa. This would not lead to heparin resistance.
Rationale: FFP may accentuate the anticoagulant effect by providing additional AT.
B. Vitamin K.
Rationale: Vitamin K is used for the reversal of warfarin.
C. Cryoprecipitate.
Rationale: Cryoprecipitate has no role in the reversal of heparin therapy.
D. Prothrombin complex concentrates.
Rationale: Prothrombin complex concentrates can be used for warfarin therapy but are not useful for heparin reversal.
E. Protamine sulfate.
Rationale: Protamine sulfate is the treatment of choice for reversal of heparin.
Rationale: The y-intercept is not used in the calculation of the ISI of a test reagent.
B. 0.90
Rationale: This is equal to the ISI + y-intercept, which is not the correct equation.
C. 0.95
Rationale: The ISI of the test reagent = ISI reference preparation × Slope of regression line.
D. 1.0
Rationale: The ISI of the test reagent would be 1.0 if the slope of the regression line were 1.0.
E. 1.1
Rationale: This is equal to the ISI of the reference preparation – the y-intercept, which is not the correct equation.
Rationale: Osteoporosis is a side effect of warfarin, but bone necrosis is not.
B. Transmissible spongiform encephalopathy.
Rationale: Warfarin is chemically synthesized; it is not derived from animals or human tissue.
C. Purple toe syndrome.
Rationale: Purple toe syndrome is a side effect of warfarin occurring approximately 8 weeks after warfarin initiation.
D. Immune thrombocytopenia.
Rationale: Heparin-induced thrombocytopenia is seen with heparin therapy.
E. Hollenhorst plaque.
Rationale: Hollenhorst plaque is composed of cholesterol emboli that most likely dislodge from an atheromatous plaque in the internal carotid and become lodged within the retinal artery; it is not an adverse consequence of warfarin therapy.
Rationale: The pre-anhepatic stage is characterized by a mild deterioration in baseline coagulopathy.
B. Anhepatic to reperfusion.
Rationale: During this stage, there is a loss of coagulation factor synthesis and clearance as well as hyperfibrinolysis, which can lead to severe bleeding.
C. Postreperfusion.
Rationale: During the postreperfusion stage, there is resolution of fibrinolysis, restoration of coagulation factor synthesis and clearance, and a heparin-like effect from the donor liver.
D. Postoperative.
Rationale: Hypercoagulability during the postoperative stage is related to the early recovery of procoagulants, elevated factor VIII, and delayed recovery of AT, protein C, and protein S.
E. During surgical dissection and mobilization of the diseased liver.
Rationale: This refers to the pre-anhepatic stage. The pre-anhepatic stage is characterized by a mild deterioration in baseline coagulopathy.
Rationale: This is the mechanism for thrombosis associated with homocysteinemia.
B. Failure to generate APC.
Rationale: This is the mechanism of thrombosis associated with protein C deficiency.
C. Failure of APC to inactivate factors Va and VIIIa.
Rationale: This is the mechanism of thrombosis associated with protein S deficiency.
D. Failure to generate plasmin.
Rationale: This is the mechanism of thrombosis associated with plasminogen deficiency.
E. Failure to inhibit factors IIa (thrombin), Xa, and other activated factors.
Rationale: Through its interaction with heparin and heparin-like glycosaminoglycans, AT III inhibits factors IIa (thrombin), Xa, and other activated factors.
Rationale: Neutralization of tPA is the mechanism of thrombosis associated with excess plasminogen activator inhibitor 1 activity.
B. Abnormal fibrinogen inhibits protein C activity.
Rationale: This is not a recognized mechanism of thrombosis caused by dysfibrinogenemia.
C. Abnormal fibrinogen resists fibrinolysis.
Rationale: This is one of two recognized mechanisms for thrombosis caused by dysfibrinogenemia.
D. Abnormal fibrinogen induces cytotoxicity and disrupts vascular hemostatic mechanisms.
Rationale: This is the mechanism of thrombosis associated with homocysteinemia.
E. Abnormal fibrinogen disrupts the activation of plasminogen.
Rationale: This is the mechanism of thrombosis associated with tPA deficiency.
Rationale: This is the mechanism for thrombosis associated with protein C deficiency.
B. Neutralization of tissue plasminogen activator (tPA).
Rationale: This is the mechanism for thrombosis associated with excess plasminogen activator inhibitor 1 activity.
C. Endothelial cell toxicity and disruption of vascular hemostatic mechanisms.
Rationale: This is the mechanism for thrombosis associated with homocysteinemia.
D. Activated protein C is unable to inactivate factor Va.
Rationale: Because of a highly conserved point mutation in factor V, APC is unable to cleave factor Va.
E. Failure to inhibit thrombin.
Rationale: This is the mechanism for thrombosis associated with heparin cofactor II deficiency.
Rationale: This is the mechanism for thrombosis in the setting of heparin cofactor II deficiency.
B. Failure to activate plasminogen.
Rationale: This is the mechanism for thrombosis associated with tPA deficiency.
C. Failure to inhibit factor IIa (i.e., thrombin), factor Xa, and other activated factors.
Rationale: This is the mechanism for thrombosis associated with AT deficiency.
D. Failure to generate APC.
Rationale: This is the mechanism for thrombosis associated with protein C deficiency.
E. Neutralization of tPA.
Rationale: This is the mechanism for thrombosis associated with excess plasminogen activator inhibitor 1 activity.
B. AT.
C. Protein S.
D. Plasminogen.
Rationale: Plasminogen is only choice produced in the liver.
E. Tissue factor pathway inhibitor.
Rationale for A, B, C, and E: TFPI is produced both in the liver and in extrahepatic sites.
B. Fibrinogen.
Rationale: Fibrinogen is synthesized only in the liver.
C. Factor V.
D. Plasminogen activator inhibitor.
Rationale for A, C, and D: Plasminogen activator inhibitor is produced both in the liver and in extrahepatic sites.
E. Plasminogen.
Rationale: Plasminogen is a fibrinolytic factor that is produced solely by the liver and is not a procoagulant factor.
Rationale: This is the mechanism of activation associated with protein C deficiency.
B. Failure to inhibit factor IIa (i.e., thrombin), factor Xa, and other activated factors.
Rationale: This is the mechanism of thrombosis associated with AT deficiency.
C. Failure to activate plasminogen.
Rationale: This is the mechanism of thrombosis associated with tPA deficiency.
D. Endothelial cell toxicity and disruption of vascular hemostatic mechanisms.
Rationale: This is the mechanism of thrombosis associated with hyperhomocysteinemia.
E. Failure of APC to inactivate factors Va and VIIIa.
Rationale: This is the mechanism of thrombosis associated with protein S deficiency.
B. There is good evidence that the PT, not the PTT, is useful in predicting bleeding or thrombotic risk in liver disease.
Rationale: The PT is not a good predictor of bleeding or thrombotic risk in patients with liver disease; in addition, the INR should not be used.
C. There is no good evidence to support the use of routine coagulation tests for predicting bleeding or thrombotic risk in liver disease.
Rationale for A and C: Routine coagulation tests are poor predictors of bleeding and thrombotic risk in patients with liver disease.
D. There is good evidence to support the use of the PTT for predicting bleeding or thrombotic risk in liver disease.
Rationale: There is no evidence that the PTT is a good predictor of bleeding or thrombotic risk in patients with liver disease.
E. There is good evidence to support the use of the thrombin time for predicting bleeding or thrombotic risk in liver disease.
Rationale: There is no evidence that the thrombin time is a good predictor of bleeding or thrombotic risk in patients with liver disease.
Rationale: This deficiency results in failure to generate APC.
B. AT deficiency.
Rationale: This deficiency results in failure to inhibit factor IIa (i.e., thrombin), factor Xa, and other activated factors.
C. Factor V Leiden.
Rationale: This variant factor V protein results in the failure of APC to inactivate factor Va.
D. Heparin cofactor II deficiency.
Rationale: This deficiency results in failure to inhibit factor IIa.
E. Excess plasminogen activator inhibitor 1 activity.
Rationale: Excess plasminogen activator inhibitor 1 activity will inhibit tPA inhibitor activity, leading to increased clot stability.
Rationale: This is the mechanism for thrombosis associated with protein S deficiency.
B. Failure to inhibit thrombin.
Rationale: This is the mechanism for thrombosis associated with heparin cofactor II deficiency.
C. Failure to generate plasmin.
Rationale: Failure to generate plasmin results in increased clot stability.
D. Failure to generate APC.
Rationale: This is the mechanism for thrombosis associated with protein C deficiency.
E. Failure of activated protein C to inactivate factor Va.
Rationale: This is the mechanism for thrombosis associated with FVL.
Rationale: This is the mechanism for thrombosis associated with AT III deficiency.
B. Failure to generate APC.
Rationale: Protein C deficiency results in a lack of APC and an inability to inactivate factors Va and VIIIa.
C. Failure to generate plasmin.
Rationale: This is the mechanism for thrombosis in plasminogen deficiency.
D. Endothelial cell toxicity and disturbance of vascular hemostatic mechanisms
Rationale: This is the mechanism for thrombosis associated with homocysteinemia.
E. Failure of APC to inactivate factors Va and VIIIa.
Rationale: This is the mechanism for thrombosis associated with protein S deficiency.
Rationale: This is the mechanism for thrombosis associated with heparin cofactor II deficiency.
B. Failure of APC to inactivate factors Va and VIIIa.
Rationale: Activated protein C requires the activity of its cofactor, protein S, to inactivate factors Va and VIIIa.
C. APC fails to inactivate factor Va because of a highly conserved point mutation.
Rationale: This is the mechanism for thrombosis associated with FVL.
D. Failure to activate plasminogen.
Rationale: This is the mechanism for thrombosis associated with tPA deficiency.
E. An acquired abnormality in fibrin results in decreased fibrinolysis.
Rationale: This is the mechanism for thrombosis associated with dysfibrinogenemia.
Rationale: This is the mechanism of thrombosis associated with hyperhomocysteinemia.
B. Failure of APC to inactivate factors Va and VIIIa
Rationale: This is the mechanism of thrombosis associated with protein S deficiency.
C. Failure to generate APC.
Rationale: This is the mechanism of thrombosis associated with protein C deficiency.
D. Failure to activate plasminogen.
Rationale: Failure to activate plasminogen to plasmin results in increased clot stability.
E. Failure to inhibit factor IIa, factor Xa, and other activated factors.
Rationale: This is the mechanism for thrombosis associated with AT deficiency.
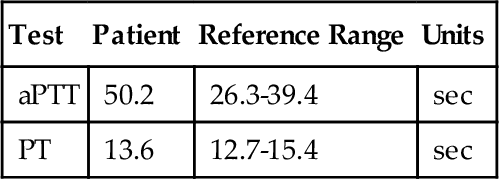
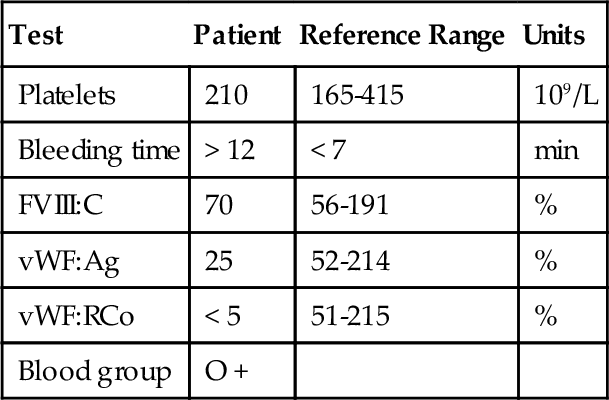
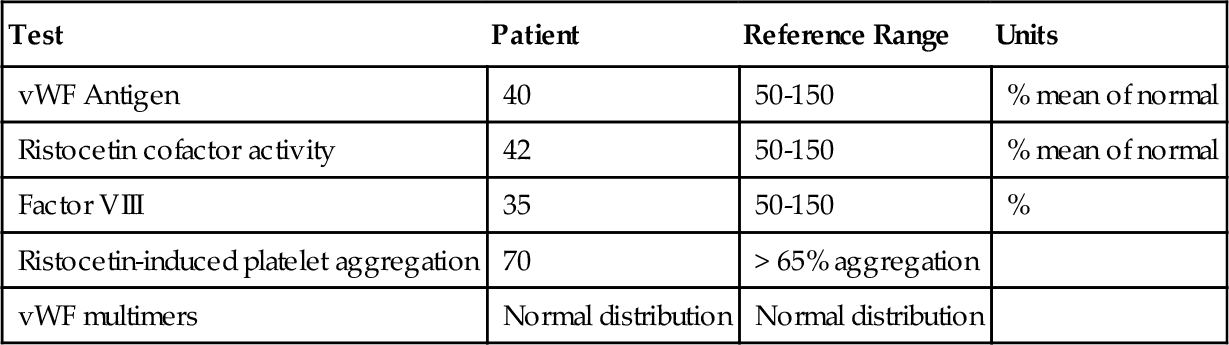
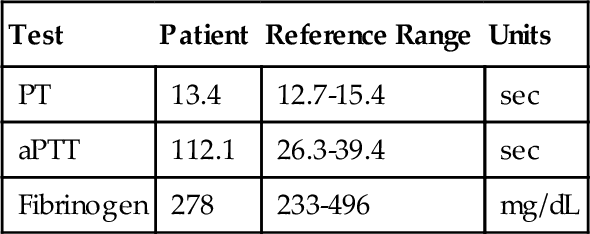
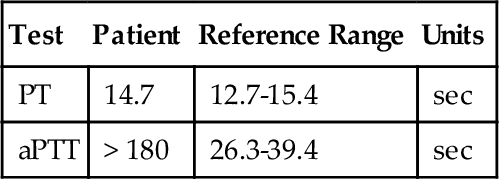
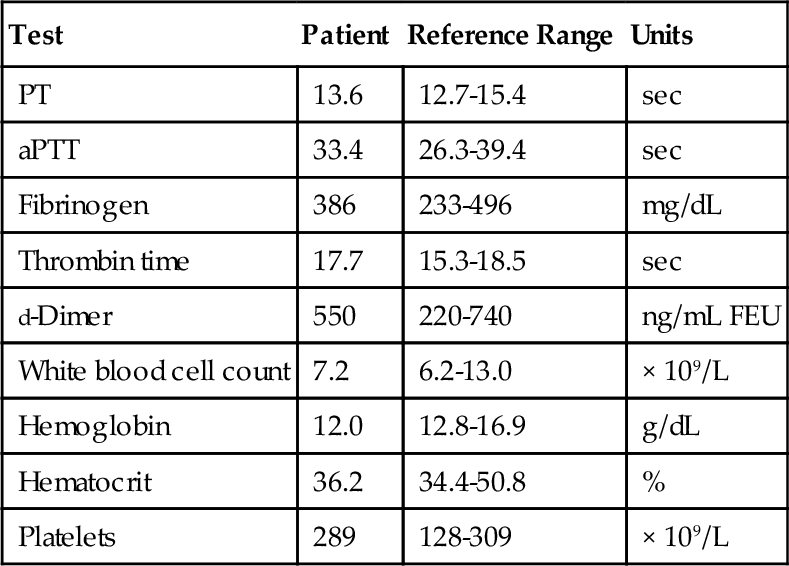
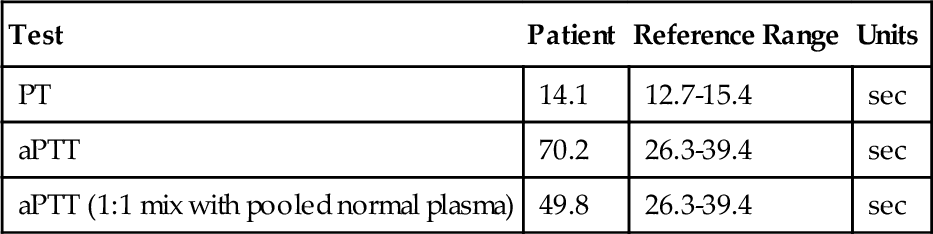
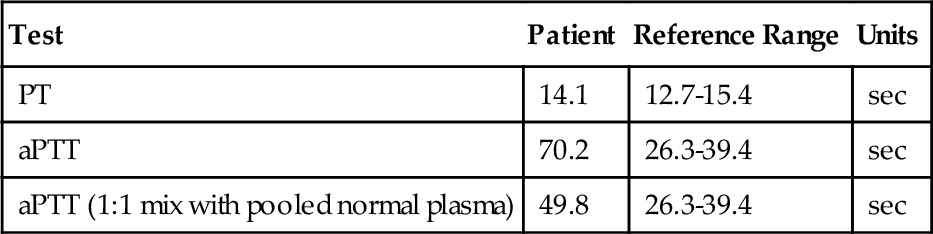
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


