Expanded Access of an Investigational Drug or an Approved Drug with Restricted Distribution

Controlled clinical trials provide the scientific and legal basis by which regulatory authorities around the world evaluate new prescription drugs and approve them for sale. In the United States, the regulatory review of drugs and devices is the responsibility of the US Food and Drug Administration (FDA). Over the past 50 years, improved methods for large-scale clinical studies have precipitated a shift toward evidence-based medicine. The increased emphasis on clinical trials to appropriately assess the safety and efficacy of new drugs has resulted in a dramatic rise in the costs associated with drug development. According to the Pharmaceutical Manufacturers of America (PhRMA), the overall drug development process from discovery to approval of a new drug takes an average of 10–15 years and an estimated cost of $1 to $2 billion; the clinical development phase of this work is typically in the range of 6 to 7 years. Moreover, only about 1 in 10 drugs that enters the clinic for testing ultimately receives regulatory approval and is marketed. Given the tremendous cost and duration of clinical drug development, it is imperative that every effort is made to plan carefully and execute effectively. Drug development programs must be well designed not only to appropriately demonstrate safety and clinical efficacy but also, through the use of appropriate biomarkers, pharmacodynamic markers, and safety monitoring, to allow for the early discontinuation of development of drugs that are destined to fail.
This is an exciting and challenging time to be involved in drug development. Major advances in the biological sciences have yielded a much greater understanding of the molecular basis of many diseases and provide the opportunity to have an unprecedented impact on the alleviation of human suffering. However, translating these scientific advances into new and more effective therapies for human diseases has proven to be daunting. In the 10 years between 1994 and 2003, there was an average of 33.6 new drugs approved per year by the FDA; in the following 10 years up to 2013, there has been an average of slightly more than 26 approvals per year, suggesting a relative stagnation in the rate of new drugs being approved and made available to patients. To close this apparent gap between the acceleration of innovative basic-science discoveries and the stagnating rate of approvals of innovative new therapies will require diligent clinical drug development programs that include rigorous, well-controlled trials, integrated clinical development plans, the use of novel statistical methods, adaptive clinical trial designs, and the inclusion of novel pharmacodynamic and other biomarkers at the various stages of drug development. Coordinated teams of experts will be needed to integrate these clinical development programs with the related processes of drug discovery, preclinical development, regulatory approval, and ultimately patient treatment. In addition, major health authorities such as the FDA have implemented programs to work more effectively with industry in drug development while preserving their remit to protect the public health.
Drug discovery and development remains a lengthy, high-risk, and complex process. It has been estimated that of every 5,000 to 10,000 chemically synthesized molecules that are screened as potential drugs, only one becomes an approved drug. The previous chapter (Chapter 51, Drug Discovery and Preclinical Development) outlines the preclinical phase of drug development from target identification to candidate selection. This chapter describes the process by which new candidate drug molecules are evaluated in clinical trials and approved for marketing and sale in the United States.
 For most of the latter half of the twentieth century, advances in the pharmacologic treatment of malignancies relied primarily on the use of cytotoxic agents that target various aspects of cellular viability and proliferation, with only a narrow window between the doses necessary for the killing of tumor cells and those that kill normal cells (i.e., the therapeutic window). In the 1970s and 1980s, studies by scientists such as Michael Bishop and Harold Varmus led to the identification of retroviral oncogenes, which are mutated forms of normal cellular genes that control cell viability, differentiation, and proliferation. Many of these oncogenes were shown to encode mutated protein kinases involved in the pathogenesis of human malignancies. Chronic myelogenous leukemia (CML) is one such malignancy that is fairly well understood at the molecular level. CML has been demonstrated to depend on a chromosomal translocation, the so-called Philadelphia chromosome, characterized by a reciprocal translocation between the long arms of chromosomes 9 and 22, which leads to the rearrangement and dysregulation of a particular tyrosine kinase called c-abl.
For most of the latter half of the twentieth century, advances in the pharmacologic treatment of malignancies relied primarily on the use of cytotoxic agents that target various aspects of cellular viability and proliferation, with only a narrow window between the doses necessary for the killing of tumor cells and those that kill normal cells (i.e., the therapeutic window). In the 1970s and 1980s, studies by scientists such as Michael Bishop and Harold Varmus led to the identification of retroviral oncogenes, which are mutated forms of normal cellular genes that control cell viability, differentiation, and proliferation. Many of these oncogenes were shown to encode mutated protein kinases involved in the pathogenesis of human malignancies. Chronic myelogenous leukemia (CML) is one such malignancy that is fairly well understood at the molecular level. CML has been demonstrated to depend on a chromosomal translocation, the so-called Philadelphia chromosome, characterized by a reciprocal translocation between the long arms of chromosomes 9 and 22, which leads to the rearrangement and dysregulation of a particular tyrosine kinase called c-abl.
These findings set the stage for an extraordinarily successful collaboration between Brian Druker, an academic oncologist at Oregon Health Sciences University, and Nick Lydon, a pharmaceutical researcher at Novartis. Druker had a research focus on tyrosine kinase biology with an emphasis on finding an effective treatment for CML by targeting the c-abl tyrosine kinase, and Lydon had a research focus on identifying specific inhibitors of protein tyrosine kinases. Druker and Lydon identified a small molecule, code-named STI-571 (imatinib), that effectively inhibited c-abl as well as at least two other tyrosine kinases, c-kit and platelet-derived growth factor receptor B. Studies in cell culture demonstrated selective toxicity of STI-571 in cells containing dysregulated c-abl, and preclinical studies in the appropriate animal models confirmed this activity. Preclinical toxicology studies in rats, dogs, and monkeys described the hematological, renal, and hepatobiliary toxicity of imatinib. A phase 1 study in 83 CML patients showed that oral dosing in the range from 25 to 1,000 mg/day did not cause dose-limiting toxicity. Additionally, the study demonstrated that imatinib had excellent oral bioavailability and a pharmacokinetic profile such that once-daily oral dosing could achieve sustained plasma levels at concentrations that had been sufficient to inhibit c-abl in preclinical models. Three open-label, single-arm phase 2 studies in 1,027 CML patients at various stages of disease progression all showed marked imatinib activity, as assessed by high cytogenetic response rates and hematological response rates, with less toxicity than that typically observed with the available standard of care (interferon-alpha).
Based on the marked activity of imatinib in patients with advanced disease and in patients who had failed first-line therapy with interferon-alpha, imatinib received accelerated approval by the FDA in May 2001, after only 3 months of review. This represented one of the fastest reviews ever performed by the FDA and also marked the approval of the first selectively targeted cancer therapy (i.e., therapy directed at a target that is specifically dysregulated in CML cells compared to normal cells). Accelerated approval was granted rather than full approval since cytogenetic responses and hematological responses are surrogate clinical endpoints thought to predict clinical benefit with a reasonable likelihood but are not an ultimate clinical endpoint such as survival. Under accelerated approval, the sponsor (in this case, Novartis) was required to conduct post-approval studies to verify and confirm the clinical benefit of imatinib. Novartis then conducted and submitted for approval a randomized phase 3 study comparing imatinib to combination therapy with interferon-alpha and cytarabine in patients with newly diagnosed CML, with a primary endpoint of overall survival. Novartis also committed to perform phase 1 and phase 2 studies of imatinib in children. Based on long-term follow-up of patients from the earlier phase 2 trials as well as new data from the phase 3 trial and the pediatric trials, imatinib ultimately received full approval for all stages of CML in both adults and children.
Questions
1. What ethical standards govern the relationship between physicians and patients in clinical research?
2. What are the critical elements to be considered in developing a clinical trial protocol?
3. What data do the FDA review when considering approval of a new drug?
 HISTORY OF US FOOD AND DRUG LAW
HISTORY OF US FOOD AND DRUG LAW
Drug development, testing, and approval is a lengthy process, the major milestones of which are shown in Figure 52-1. Achievement of each of these milestones requires the cooperation of researchers, clinicians, patients, pharmaceutical or biotechnology companies, and government regulators. The development of a new drug or biotherapeutic is a highly regulated process that has evolved considerably during the last century. Several public health crises have led to the development of current laws and regulations, including:
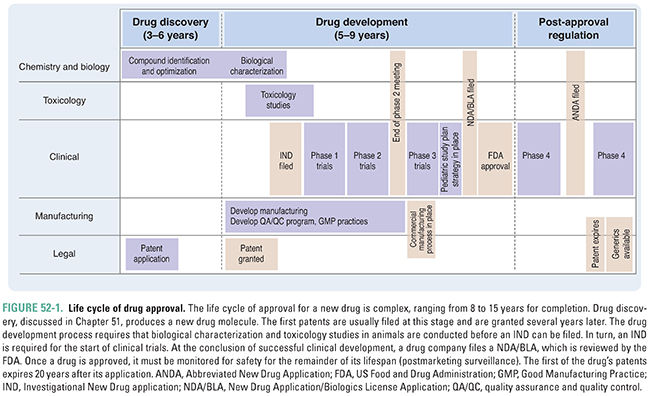
1. The public outcry over the unsanitary and unsafe conditions of the meatpacking industry: this resulted in the Pure Food and Drugs Act in 1906, which prohibited interstate commerce in adulterated and misbranded food and drugs.
2. The death of more than 100 people after consuming “Strep-Elixir,” an untested product containing a sulfonamide and a chemical analogue of antifreeze: this resulted in the passage of the Food, Drug, and Cosmetic Act in 1938, which, among other things, required approval of all new drugs by the FDA prior to marketing. Sponsors submitted an application for approval and, unless the FDA determined that the drug was unsafe within 180 days, the drug could be marketed.
3. The discovery that thalidomide, used to treat morning sickness, caused birth defects in large numbers of babies born in Europe: this resulted in passage of the Kefauver-Harris Amendments in 1962, which required proof of efficacy in addition to safety prior to drug approval and mandated reporting of adverse events. Additionally, the Amendments stated that patients must provide informed consent to participate in clinical trials, gave the FDA authority to regulate prescription drug advertising, and introduced good manufacturing practice standards.
4. In response to widely publicized safety issues with COX-2 inhibitors and several other drugs, Congress passed the FDA Amendments Act (FDAAA) of 2007. One aspect of the FDAAA provides enhanced authority to the FDA to manage the safety of approved drugs. In particular, the FDA has focused on the implementation of Risk Evaluation and Mitigation Strategies (REMS) for selected new drugs as well as drugs that are already approved. The objective of REMS is to put in place measures to ensure that a drug or biologic product is dispensed and utilized in such a manner as to ensure that its benefits outweigh its risks.
5. The FDA Safety and Innovation Act (FDASIA) of 2012 expanded FDA authority in several ways. It reauthorized prescription drug and medical device user fees and implemented generic drug and biosimilar biologic products user fees. These fees are mandated by law and are used, in part, to support the FDA’s review of marketing applications. FDASIA also introduced a new mechanism to expedite development and review of promising new drugs for serious and life-threatening diseases. These drugs are given Breakthrough Therapy designation. This mechanism builds on past FDA programs to assist sponsors in drug development. Additionally, FDASIA expands the FDA’s ability to obtain patient input into the drug development and review process and, recognizing the increasing globalization of drug supply and sourcing (for both finished product and active ingredients), it expands FDA authority to ensure the safety and availability of this supply.
In the United States, the FDA Center for Drug Evaluation and Research (CDER) and the FDA Center for Biologics Evaluation and Research (CBER) are responsible for regulating the development and approval of new medicines.
 ETHICS IN CLINICAL DRUG INVESTIGATION
ETHICS IN CLINICAL DRUG INVESTIGATION
The development of new therapeutics to treat human diseases requires research to be conducted on human subjects, either normal volunteers (typically in phase 1 trials) or in patients with the disease for which the new treatment is being investigated. Any time research is conducted in humans, it is essential that every effort is made to protect their safety. Regulatory agencies around the world have codified standards of ethical behavior for all parties involved in clinical research, including clinicians, pharmaceutical companies, and medical institutions. The ethical relationship is governed by the notion that clinical trial research represents a partnership between investigator (physician) and subject (volunteer or patient). Four major ethical principles, established by the International Conference on Harmonization and the Declaration of Helsinki, support this partnership. These principles are as follows:
 The trial must minimize the risks for participants.
The trial must minimize the risks for participants.
 Provisions must be made for the overall care of the patient.
Provisions must be made for the overall care of the patient.
 The investigator is responsible for terminating the trial when the risks become incompatible with the goals of the trial.
The investigator is responsible for terminating the trial when the risks become incompatible with the goals of the trial.
 Adverse events must be reported immediately to an ethics or safety committee.
Adverse events must be reported immediately to an ethics or safety committee.
Investigators must obtain subjects’ informed consent. Informed consent is not just a signed document but rather a process in which patients (1) are made aware of the potential risks and benefits of the trial and (2) must make an informed decision to participate voluntarily in a clinical study. For patients with poor prognoses and for normal volunteers, informed consent encompasses the understanding that the research likely will not benefit them but may benefit future patients.
At the institutional level, the FDA relies on independent Institutional Review Boards (IRBs) or Independent Ethics Committees (IECs) to ensure the rights and welfare of those participating in clinical trials. FDA regulations mandate that clinical study protocols be reviewed for legal and ethical issues by an IRB/IEC. These regulations give IRBs/IECs the authority to approve, require modification of, or disapprove research on human subjects. Specifically, the IRB/IEC must determine whether the proposed research:
 Minimizes potential risk to human subjects
Minimizes potential risk to human subjects
 Poses risks that are reasonable relative to the anticipated benefit and potential scientific gain of the research
Poses risks that are reasonable relative to the anticipated benefit and potential scientific gain of the research
 Includes equitable selection of subjects
Includes equitable selection of subjects
 Provides for an effective informed consent process
Provides for an effective informed consent process
 Contains safeguards for vulnerable populations, such as children and the mentally disabled
Contains safeguards for vulnerable populations, such as children and the mentally disabled
IRB/IEC oversight and approval begins before the commencement of human trials and continues for the duration of clinical trials. The membership of an IRB/IEC consists of five or more experts and laypersons from various backgrounds. Federal regulations stipulate that IRB membership must include at least one member whose primary expertise is in a scientific area, one member whose primary expertise is in a nonscientific area, and one member who is not affiliated with the institution overseeing the clinical research protocol. In addition, the other members’ qualifications must be such that the IRB is able to evaluate research protocols in terms of institutional requirements, applicable law, standards of professional practice, and community attitudes. Thus, many IRBs include clergy, social workers, and attorneys as well as physicians, scientists, and other health care professionals.
Clinical trials must be appropriately designed and rigorously executed in order to optimize the ratio of benefit to risk and to satisfactorily answer the scientific questions under study. Scientific clinical trial design must include appropriate control or comparator arm(s), randomization and blinding, and sample size, among other elements (see below). Some institutions have a scientific review committee that must approve all protocols involving human subjects to ensure that the protocol is appropriately designed to answer the questions being asked. To further assure that the findings of clinical trials are accurate and credible and that the rights of clinical trial subjects are protected, regulatory agencies require that clinical trials leading to the approval of new drugs be conducted according to good clinical practices (GCP). Guidelines for GCP have been developed by the International Conference on Harmonization to provide a standard for the design, conduct, recording of findings, monitoring of data, analysis, auditing, and reporting of results of clinical trials.
 DRUG EVALUATION AND CLINICAL DEVELOPMENT
DRUG EVALUATION AND CLINICAL DEVELOPMENT
The investigation of a new drug candidate comprises several phases, beginning with preclinical evaluation and typically proceeding through phase 3 clinical studies. At the conclusion of this process, the FDA may consider the molecule for approval as a new drug.
Authorizations to Initiate Clinical Trials
Preclinical research and development establishes the potential efficacy and safety of a compound for use in human trials. During this stage of testing, described in Chapter 51, a compound is studied to determine its biological actions, chemical properties, and metabolism, and a process is developed for its synthesis and purification. A major focus of preclinical testing is determining whether the molecule has an acceptable safety profile in animals prior to initiating testing in humans. The International Conference on Harmonization has established requirements for the animal studies used to support different types of clinical trials. The primary studies used to support clinical drug development are animal toxicity studies and investigations on the absorption, distribution, metabolism, and excretion (ADME) of the compound. As described in Chapter 51, the duration of animal studies is determined by the length of the clinical trials to be undertaken. For this and other reasons, it is essential that there is close coordination among the preclinical and clinical scientists on the drug development team. Many potential drug candidates either do not proceed to human trials or are removed from clinical testing due to adverse safety findings in animal studies. The preclinical research phase is also an important time to explore potentially important pharmacodynamic markers and other biomarkers that could help facilitate clinical development.
The mechanism for seeking approval to initiate clinical trials in the United States is the submission of an Investigational New Drug application (IND) to the FDA. The IND contains data from the preclinical studies, data from prior clinical investigations (if available), the proposed protocol for human trials, and other background information. The IND also contains a document referred to as the Investigator’s Brochure (IB). The IB is provided to regulators, clinical investigators, and IRBs/IECs; it represents a summary of all available information on the investigational drug and may be several hundred pages in length. The IND must also contain information on characterization, manufacture, and quality of the drug. INDs can be submitted by commercial sponsors with the ultimate goal of obtaining approval for marketing and sale of a new drug product or by individual investigators and academic centers. The latter are typically referred to as Investigator Sponsored INDs. The IND is a “living document” and, at a minimum, is updated annually.
The FDA must review the IND within 30 days and decide whether human trials may begin. Figure 52-2 is a flowchart representing the process used by the FDA to review an IND. The areas of review include a chemistry review, a pharmacology/toxicology review, and a medical review. If the IND review does not identify any safety concerns, the IND is considered open or active after the 30-day wait period. If the review reveals the potential for unreasonable risk to participants, the FDA contacts the sponsor, and a clinical hold is issued, preventing initiation of human studies. The sponsor must address any issues in question before the clinical hold is lifted. A clinical hold may be issued at any time during clinical drug development; this can be based on issues such as new findings from animal studies, clinical data indicating an unacceptable risk profile, or a finding that a sponsor did not accurately disclose the risk of the study to investigators or subjects.
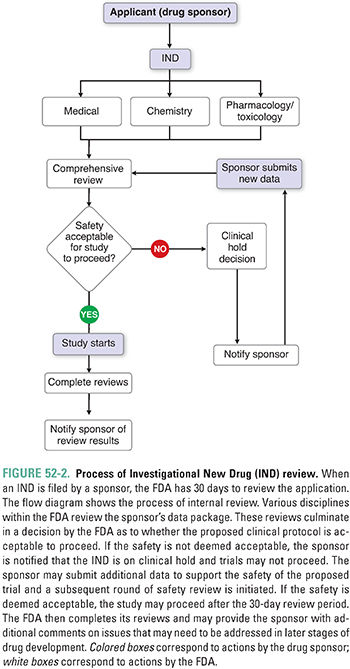
Given the time, cost, and risks associated with clinical drug development, it is imperative to plan carefully and execute meticulously. The goals of clinical drug development include:
 Assessment of the dose–response profile
Assessment of the dose–response profile
 Assessment of the toxicity profile for a given dosing regimen
Assessment of the toxicity profile for a given dosing regimen
 Assessment of pharmacokinetic/pharmacodynamic relationships
Assessment of pharmacokinetic/pharmacodynamic relationships
 Establishment of the safety and efficacy profile in well-controlled studies in well-defined patient populations
Establishment of the safety and efficacy profile in well-controlled studies in well-defined patient populations
These goals are accomplished through the conduct of clinical trials. Each clinical trial must be designed to answer specific questions. In turn, each trial should be part of an integrated development plan leading to the ultimate demonstration of safety and efficacy in well-controlled trials.
The target product profile (TPP) articulates the goal of the clinical development program. The key elements of the target product profile include the primary indication, target patient population, route of administration, pharmaceutical formulation, dosing schedule, efficacy assessments, expected primary endpoint in pivotal trial(s), expected safety profile, and key product characteristics, including those that may allow differentiation compared to products already available. As the development plan unfolds, data from clinical trials and product development inform the TPP and, as a result, aspects of the TPP are likely to evolve. However, it is important to understand what the minimally acceptable target product profile is and to have the discipline to terminate development as quickly and responsibly as possible if it becomes clear that the minimally acceptable TPP will not be attainable.
Development of a Clinical Trial
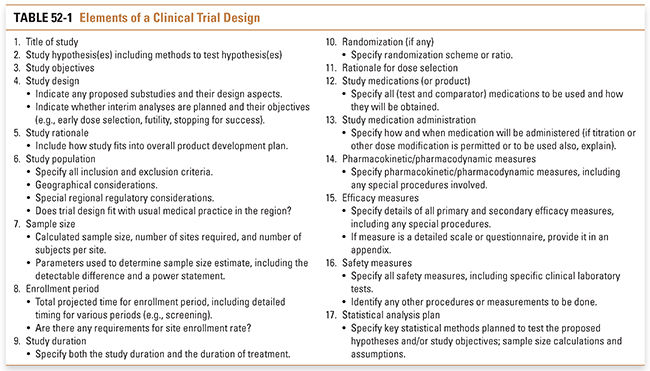
By definition, clinical trials involve studies of human subjects. The subjects may be normal volunteers or patients with specific diseases; the trials may be interventional (i.e., patients receive therapies and/or undergo tests or procedures) or observational (e.g., a study of the natural history of a disease). No matter what the situation, the clinical investigator has an ethical responsibility to the subjects to ensure that all elements of a trial are optimally designed to maximize what will be learned. The key elements for consideration when developing any clinical trial are detailed in Table 52-1.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


