Figure 59-1. Evolution of cirrhosis. Fibrosis develops in nonregenerative necrotic areas, producing scars. The pattern of nodularity and scars reflects the type of response to injury (e.g., uniform vs. nonuniform necrosis) and the extent of injury.
Alcoholic liver disease has long been known to be associated with consequences of oxidative stress in the liver with failure of homeostatic mechanisms.11 Obesity and metabolic syndrome, a major health problem in the United States, may produce hepatic injury and may potentiate the effects of viral injury.12,13 In addition to direct oxidative stress, hepatocyte injury is mediated by a variety of mechanisms including proinflammatory cytokines1 and failure of reparative or modulatory pathways.14
The failure of protective or reparatory mechanisms is also widely studied.15 Over time, fibrosis is the inexorable consequence of ongoing hepatic injury. HSCs are located in the perisinusoidal space of Disse (Fig. 59-2).16 In the normal liver, these cells are quiescent and are primarily responsible for the storage of vitamin A.17 Injured hepatocytes release soluble factors that activate HSCs to differentiate into myofibroblastic HSCs, as evidenced by cellular enlargement and proliferation, an increase in rough endoplasmic reticulum, loss of vitamin A droplets, expression of actin filaments, and increased expression of “fibril-forming” collagen types I, III, and V.18 They also express components of the extracellular matrix, including heparan sulfate, dermatan, chondroitin sulfate,19 laminin,20 and fibronectin.21 Kupffer cells secrete TGF-β1, which appears to be critical for the activation of HSCs.5,22 Both TGF-β and PDGF have been shown to enhance proliferation and fibrogenesis in animal models,5,7,23 with TGF-β being the primary stimulator of collagen synthesis and fibrosis. Further evidence implicating TGF-β in the production of hepatic fibrosis is the observation that levels of TGF-β are reduced by therapy with interferon-α in patients who are positive for hepatitis C. This reduction has been correlated with a regression of hepatic fibrosis.24
CLASSIFICATION
Table 59-1 Classification of Cirrhosis

In addition to the well-characterized effects of TGF-β1 and PDGF on activating HSCs, more recent research has demonstrated the central role of Hedgehog (Hh) in activation of HSCs and promotion of fibrosis.25 Hh is a molecule involved in signaling pathways that help determine cell fate during embryogenesis. The Hh ligand binds to its cell surface receptor patched. Binding of Hh to patched releases another effector, named smoothened, from a chronic state of inactivation. Activation of smoothened leads to downstream activation of a number of proteins involved in nuclear transcription, thereby leading to changes in cell fate and differentiation.25 In the liver, the two primary forms of Hh are Sonic Hedgehog (SHh) and Indian Hedgehog (IHh), which are expressed by hepatocytes, bile duct cells, and HSCs. In the healthy liver, quiescent HSCs and endothelial cells keep Hh inactive by production of inhibitory proteins. When the liver is injured, however, numerous cell types including hepatocytes, cholangiocytes, HSCs, progenitor cells, lymphocytes, duct cells, and endothelial cells produce elevated levels of Hh under the influence of cytokines such as PDGF and TGF-β.25–30 Activation of the Hh pathway tends to be self-sustaining and resistant to negative feedback as long as the injury process continues.31 It is this surge in Hh levels within the local hepatic milieu that signals quiescent HSCs to activate. The critical role of Hh signaling in HSC activation and fibrogenesis was recently demonstrated by Michelotti et al.,32 who showed that deletion of the Hh intermediary smoothened was sufficient to inhibit both HSC activation and fibrosis formation.

Figure 59-2. Matrix and cellular alterations in hepatic fibrosis. A: In normal liver, a modest amount of low-density matrix is present in the subendothelial space of Disse. B: In the fibrotic liver, the accumulation of fibril-forming matrix in this region leads to “capillarization” of the sinusoid and functional changes in all neighboring cell types.
Another interesting concept that has been gaining increasing recognition is the role of the gut microflora in the development of cirrhosis. Alcoholics, for example, are known to have a greater susceptibility to small intestinal bacterial overgrowth along with alterations in their gut microflora. This bacterial overgrowth and dysbiosis lead to increased levels of LPS, which reach the liver to stimulate Kupffer cells to produce proinflammatory cytokines. The end result is the generation of mitochondrial damage, reactive oxygen species, and ongoing hepatic inflammation.33–35 A similar role for alterations in gut microflora has been demonstrated in nonalcoholic fatty liver disease (NAFLD).36,37
As a result of the activation of stellate cells and a subsequent enhancement in collagen and extracellular matrix synthesis, the space of Disse becomes thickened, so that “capillarization” develops and the normal fenestrated architecture of the sinusoidal endothelium is lost.38 Obliteration of sinusoidal fenestrations may be the essential component of fibrosis-induced hepatocellular dysfunction in cirrhosis, preventing the normal flow of nutrients to hepatocytes and increasing vascular resistance.39 In addition, production of endothelin-1, a potent vasoconstrictor, by endothelial or stellate cells can cause contraction of the myofilaments within the stellate cell, influencing blood flow to injured areas and contributing to portal hypertension.40 Initially, fibrosis may be reversible if the inciting agents are removed. With sustained injury, the process of fibrosis becomes irreversible and leads to cirrhosis. Growing attention has been given to approaches that might disconnect hepatic injury from the inexorable path of fibrosis.41
Among the most promising agents currently receiving the greatest attention in the treatment of fibrosis are the antioxidants.4 The combination of N-acetylcysteine and metformin taken for 12 months has demonstrated reduction of fibrosis severity in patients with nonalcoholic steatohepatitis (NASH).42 The glutathione donor S-adenosylmethionine (SAMe) has demonstrated improved mortality in alcoholic cirrhosis, and vitamin E has been shown to improve hepatic fibrosis in the setting of NAFLD.4 While the results of studies utilizing these agents have had mixed results, ongoing investigations should establish their role in the treatment of fibrosis. Other studies have focused on utilization of inducible pluripotent stem cells and embryonic stem cells to replenish functional hepatocytes and restore liver function. Mesenchymal stem cells have been studied for their ability to reduce the profibrotic and proinflammatory milieus in the setting of hepatic damage. While these studies are in early stages, their potential is exciting.43,44
Classification Systems
Morphology
In 1977, the World Health Organization divided cirrhosis into three categories based on the morphologic characteristics of hepatic nodules (Fig. 59-3).45
Micronodular Pattern. Nodules are almost always less than 3 mm in diameter, are relatively uniform in size, are regularly distributed throughout the liver, and rarely contain portal tracts or efferent veins. Micronodular livers are usually of normal size or are mildly enlarged, and the fibrous septa vary in thickness. These changes reflect relatively early disease and are characteristic of a wide range of disease processes, including alcoholism, biliary obstruction, venous outflow obstruction, hemochromatosis, and Indian childhood cirrhosis.
Macronodular Pattern. In this category, nodules vary considerably in size and are larger than 3 mm in diameter, with some nodules measuring several centimeters. Portal structures and efferent veins are present but display architectural distortion. These livers are usually coarsely scarred with variably thick and thin septa and may be either normal or reduced in size. Two separate subcategories are recognized based on the nature of the fibrous septa. In the first category, characteristic of “posthepatitis” pathology and found in Wilson disease, fine, sometimes incomplete septa link portal tracts; these are difficult to see on gross inspection of the liver. The second is characteristic of “postnecrotic” disease, commonly found in patients with viral hepatitis, and is characterized by coarse, thick septa that are readily apparent on gross examination. Because of the relatively large size of the nodules relative to the size of biopsy specimens, diagnosis by biopsy may be difficult in macronodular cirrhosis.

Figure 59-3. A: Small, shrunken liver and a fairly regular pattern of nodularity. This appearance is rather typical of end-stage cirrhosis, regardless of the cause. B: Photomicrograph of cirrhotic liver tissue, showing irregular nodules of regenerating hepatocytes surrounded by scar. Trichrome stain. (From Stal P, Broome U, Scheynius A, et al. Kupffer cell iron overload induces intercellular adhesion molecule-1 expression on hepatocytes in genetic hemochromatosis. Hepatology 1995;21:1308–1316.)
Mixed Pattern. This description is applied to livers in which both micronodules and macronodules are present in approximately equal proportions.
Etiology
Another commonly used method for classifying cirrhosis is by etiology. The causes of cirrhosis and the morphologic and histologic characteristics of the liver, however, overlap significantly. Oxidative stress leading to chronic injury and inflammation appears to be a common theme of these disorders, which leads to both scar formation and an increased risk of liver cancer.
Alcohol. The relationship between alcohol and liver disease has been well established. In 1849, Rokitansky, referring to the association of alcohol intake and liver disease, coined the term Laennec cirrhosis.46 Consumption of at least 30 g of alcohol per day in women and 50 g of alcohol per day in men over at least 5 years is considered to the minimum threshold alcohol intake for cirrhosis to develop.47 More than 50% of alcoholics with cirrhosis and two-thirds of patients with alcoholic hepatitis and cirrhosis die within 4 years of diagnosis.48 Alcoholic cirrhosis is the second leading indication for liver transplantation overall, accounting for 40% of liver transplants in Europe and 25% in the United States.49–51 Cirrhosis, however, develops in only 10% to 30% of heavy drinkers.52 The reasons why cirrhosis develops in some alcoholics but not in others are not clear and may depend on a variety of factors, such as genetic predisposition, nutritional effects, concomitant drug use, and viral infection.
Alcoholic liver disease usually begins with a transition of normal architecture to fatty liver and alcoholic hepatitis, indicated histologically by the presence of megamitochondria, Mallory bodies (eosinophilic accumulations of intermediate filaments with cytokeratin proteins), inflammation and necrosis, and ultimately fibrosis (Fig. 59-4). Classically, the morphology of alcoholic cirrhosis is a micronodular pattern.
2 Although alcohol may directly activate stellate cells to produce collagen independently of inflammation and necrosis,53 the key mediator in alcohol-induced liver disease is acetaldehyde, the product of alcohol metabolism by the enzyme alcohol dehydrogenase. Acetaldehyde (ADH) produces numerous deleterious effects on the liver, including the following: direct activation of stellate cells54; inhibition of DNA repair55; depletion of glutathione, which impairs mitochondrial function and the ability to handle free radical production; damage to microtubules, which causes protein and water sequestration52; and formation of reduced nicotinamide adenine dinucleotide (NADH), which opposes gluconeogenesis and inhibits fatty acid oxidation, so that steatosis and hyperlipidemia develop.52 ADH is most active in the perivenular/centrilobular zone 3 of the hepatic lobule; as a result, relatively high concentrations of acetaldehyde are found in this area of the liver. In addition, zone 3 is hypoxic because of its distance from portal venous and hepatic arterial inflow. These two factors are presumably responsible for the characteristic initial perivenular location of alcohol-induced liver disease.

Figure 59-4. Alcoholic hepatitis. Mallory bodies (arrows) are evident within the swollen, clear cytoplasm of several hepatocytes. This hyaline material is chemotactic for leukocytes, many of which are seen within the field. Hematoxylin and eosin (H&E) stain × 470.
Other effects of ADH include induction of lipid peroxidation with subsequent loss of integrity of cell membranes, which causes the characteristic “ballooning degeneration” of alcohol-induced liver disease. In addition to its direct hepatic effects, ADH is now known to play a major role in the derangement of the gut–liver axis which plays a key role in alcohol-induced liver injury.56 In the “leaky gut hypothesis,” ADH increases the permeability of the intestinal barrier,57 allowing bacterial endotoxin (LPS) access to the liver via the portal circulation. Excess circulating endotoxin then activates Kupffer cells through interaction with Toll-like Receptor 4 (TLR4) to set off the inflammatory cascade responsible for the development of alcoholic liver disease.35 Necrosis and inflammation in the perivenular region activate the stellate cells in the space of Disse, so that fibrosis develops. With continued ingestion of alcohol and hepatic injury, expansion of the areas of fibrosis toward the periportal regions leads to bridging fibrosis and ultimately cirrhosis.
3 Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. As noted earlier, this entity has become a major health problem in the United States and adds even further to the litany of health consequences of obesity. NASH, as this disease was previously called, is only one stage in the NAFLD process.58 NAFLD is now the most common cause of chronic liver disease in the United States, affecting up to 30% to 46% of the population.59,60 NASH-related cirrhosis is expected to surpass viral hepatitis as the leading indication for liver transplantation by 2025.25 It is characterized by infiltration of the liver with fat, with or without inflammation (hepatitis), which shares pathologic features of alcohol-induced liver injury but occurs in patients who do not abuse alcohol. NAFLD is associated with obesity, hyperlipidemia, cardiovascular disease, and noninsulin-dependent diabetes, with 90% of NAFLD patients having at least one of these risk factors and 30% having three or more.61 It now appears that most patients traditionally diagnosed with cryptogenic cirrhosis have NAFLD.62 In addition to liver injury and cirrhosis, NAFLD is a major risk factor for primary liver cancer HCC and, in addition to hepatitis virus infection, accounts for the rapid rise in the incidence of HCC in Western countries.63 More broadly considered, recent reports have identified obesity as a major risk factor for cancer deaths with a 4.5-fold increase of the relative risk for liver cancer in obese subjects.64
The current understanding of NAFLD and progression to NASH and cirrhosis involves a two-hit model, which is nicely explained in a review by Bohinc and Diehl.25 In this model, insulin resistance leads to hepatic steatosis (the first hit), which sensitizes the hepatic parenchyma to environmental and extracellular insults (the second hit) leading to inflammation, necrosis, and fibrosis.25 Dietary fructose has been identified as an important culprit in the development of hepatic steatosis by decreasing intracellular ATP and transforming to lipid in the absence of insulin.65 Ongoing hepatic insults lead to activation of the inflammasome, a protein scaffold that results in the secretion of the inflammatory cytokines IL-1β and IL-18, further amplifying hepatic inflammation and injury. Inflammasome activation is increased by gut-derived bacterial endotoxin. This inflammation promotes increased Hh signaling and stellate cell activation with resultant fibrosis.66
Therapy for NASH centers on lifestyle modification, with a modest weight loss of 5% to 10% associated with significant histologic improvements in disease severity.61 Now that the role of gut microflora is understood to play a major role in the pathogenesis of NASH and other liver diseases, the use of probiotics, most commonly lactobacilli and bifidobacteria, is being explored in the treatment of NASH. A recent meta-analysis of four randomized controlled trials demonstrated probiotics significantly decreased transaminases, total cholesterol, TNF-α, and insulin resistance.67
4 Hepatitis. Viral hepatitis is the most common cause of cirrhosis worldwide, accounting for at least 50% of cases. Hepatitis A, B, C, D, and E have all been proved to cause acute hepatitis, characterized histologically by lymphocytic parenchymal and portal inflammations, focal necrosis, ballooning degeneration, cholestasis, Kupffer cell and macrophage hypertrophy and hyperplasia, and lobular disarray. Only hepatitis B, C, and D have been shown to progress to chronic hepatitis, defined by persistent liver cell necrosis and inflammation lasting longer than 6 months. Chronic infection with hepatitis B virus (HBV) develops in fewer than 5% of patients who experience acute HBV infection. The development of cirrhosis in approximately 10% to 20% of chronically infected persons produces an overall rate of cirrhosis of approximately 1%. Hepatitis B remains a major public health problem in Asian countries and Africa, with up to 10% of the population showing evidence of chronic infection. The widespread use of antiviral medications effective against hepatitis B effectively suppresses viral replication to undetectable levels in up to 90% of patients, and may reverse fibrosis and even cirrhosis. Even with effective suppression of viral replication, however, HBV is not truly “cured” since the virus is incorporated into the host genome. This has important consequences as HCC can still develop in the noncirrhotic liver of HBV-infected patients.68 In addition, public health strategies with systematic vaccination of newborns should substantially decrease the incidence of new infections.
In contrast, hepatitis C poses a much higher risk of chronic infection, and therapeutic strategies eradicate viral replication in a minority of patients. Of patients with hepatitis C virus (HCV), 90% become chronically infected, and chronic hepatitis develops in 60% of these patients. Among patients with chronic hepatitis, 30% progress to cirrhosis,46 so that the incidence of cirrhosis in patients initially infected with hepatitis C is approximately 10%. Standard treatment for HCV was previously based on the combination of pegylated interferon and ribavirin, with sustained viral response (SVR) rates of up to 80% in patients with genotypes 2 and 3,69 but results for the more common genotype 1 were only 40% to 50%, with the best reporting an SVR rate of 65%.70 Compounding the limited efficacy of interferon-based therapy is the high rate of adverse events that prevents many patients from completing therapy.
The recent release of a new wave of oral antiviral therapies, termed direct-acting antivirals (DAAs), and allowing interferon-free regimens, however, has demonstrated more promising results (Table 59-2).
The fixed dose combination of 90-mg ledipasvir and 400-mg sofosbuvir is sold under the trade name Harvoni (Gilead Sciences) and is recommended as first-line therapy by the American Society for the Study of Liver Disease (AASLD) for the treatment of HCV genotypes 1a, 1b, and 4.71 Ledipasvir is an inhibitor of the HCV protein NS5A, which limits viral replication. Sofosbuvir is a nucleotide inhibitor of the NS5B polymerase that terminates viral replication. In the ION-1 trial that leads to approval of the combination of these two agents for the treatment of HCV genotype 1, SVR rates of 97% to 99% were demonstrated after 12 weeks of therapy. No patients who received the 12-week regimen had adverse effects leading to early treatment discontinuation.72 For treatment of genotype 4, the SYNERGY trial demonstrated an SVR rate of 95% by intention to treat analysis following 12 weeks of ledipasvir/sofosbuvir therapy.71 Ledipasvir/sofosbuvir is also recommended as first-line therapy for genotype 6. Sofosbuvir also has significant activity against genotypes 2 and 3 when combined with ribavirin, and genotypes 5 and 6 when combined with ribavirin and pegylated interferon.71 Simeprevir is a novel NS3/4A protease inhibitor recommended as first-line therapy for HCV for genotypes 1a and 1b when used in combination with sofosbuvir. In the COSMOS trial, combination simeprevir/sofosbuvir for genotype 1 HCV demonstrated 95% SVR after 12 weeks of therapy in noncirrhotics, and 100% SVR after 24 weeks of therapy in cirrhotics.73
ETIOLOGY
Table 59-2 Causes of Cholestasis

Another novel all-oral HCV regimen is the combination of ombitasvir, paritaprevir, ritonavir, and dasabuvir, marketed as the Viekira Pak (AbbVie). Ombitasvir inhibits NS5A, paritaprevir inhibits the NS3/4A protease, ritonavir is a CYP3A4 inhibitor without any HCV activity used to increase sensitivity to paritaprevir, and dasabuvir is a nonnucleotide NS5B polymerase inhibitor. The combination is recommended as first-line therapy for genotype 1a when used with weight-based ribavirin, genotype 1b (with ribavirin for cirrhotics), and genotype 4 (with ribavirin).71 Against HCV genotype 1, SVR rates of 95.3% to 97% have been demonstrated.74,75
These results represent a revolution in the therapy of HCV that makes cure of HCV a possibility. In patients who respond to therapy, progression to cirrhosis is eliminated and it is generally thought that HCC does not develop. In patients who do not respond to therapy, cirrhosis develops in approximately 40%, and HCC develops in 16% of these patients.73
Hepatitis D virus (HDV) is an RNA virus that requires the presence of HBV to be pathogenic. Superinfection of HBV-positive patients with HDV leads to a more rapid clinical course, with progression to cirrhosis in 70% to 80% of patients.74 Among patients with compensated cirrhosis of viral origin, HCC developed in approximately 20% with HCV, 9% with HBV, and 41% with both HBV and HCV.46 The progressive increase in hepatitis-infected individuals in the United States precedes an epidemic in HCC, which has been well documented and is expected to peak over the coming decade.75
Cholestasis. Cholestasis, defined as a decrease or absence of bile flow into the duodenum, may be caused by intrahepatic or extrahepatic biliary obstruction or defects in the ability of hepatocytes to excrete bile. Causes of cholestasis are presented in Table 59-2. Prolonged biliary obstruction leads to proliferation of bile ducts, formation of bile lakes caused by disruption of bile ducts, fibrosis, and ultimately secondary biliary cirrhosis as a result of the direct toxic effects of bile salts on hepatobiliary elements.
Immune or Inflammatory Cirrhosis. Although cholestasis is a generically toxic insult to the liver that can cause cirrhosis, several diseases that are characterized by cholestasis seem to be caused by underlying immune or inflammatory disorders affecting either small (primary biliary cirrhosis [PBC]) or large (primary sclerosing cholangitis [PSC]) bile ducts. PBC is a chronic, slowly progressive disease that most commonly affects middle-aged women; it is characterized by portal inflammation, destruction of intrahepatic bile ducts, and progression to cirrhosis. Approximately 95% of patients are women, and 95% of these women express antimitochondrial antibodies in serum.76 The autoimmune inflammatory process damages both the bile ducts and, eventually, the hepatocytes as a result of leakage of bile acids into surrounding parenchyma.77 Patients present with fatigue, jaundice, and pruritus, but as many as 60% of patients may be asymptomatic.78 PBC progresses in the majority of patients; median survival times are approximately 10 to 15 years in asymptomatic patients and 7 years in symptomatic patients.79,80 Poor prognostic factors include hyperbilirubinemia, advanced age, hepatosplenomegaly, and symptomatic disease.
Medical management of PBC is with the exogenous bile acid ursodeoxycholic acid (13 to 15 mg/kg/d). Complete response, as evidenced by normalization of liver enzymes without histologic progression, is seen in up to 30% of patients and can even lead to a normal life expectancy. Therapy for nonresponders includes immunosuppression with methotrexate, colchicine, or steroids, although the efficacy of these medications is in question.81 Liver transplantation is ultimately required as the definitive treatment in most patients, and the parameters for liver transplantation and its timing are relatively predictable and have been well studied.82
PSC is a chronic, progressive cholestatic liver disease of unknown cause characterized by diffuse segmental intrahepatic or extrahepatic biliary ductular strictures with associated fibrosis and inflammation. The disease has no cure and often leads to secondary biliary cirrhosis, portal hypertension, hepatic failure, and cholangiocarcinoma if hepatic transplantation is not performed. PSC is strongly associated with inflammatory bowel diseases, most commonly ulcerative colitis, and probably shares an underlying disturbance related to disturbed mucosal immunity of the gastrointestinal tract.83 Approximately 70% of patients with PSC also have ulcerative colitis.83 Conversely, approximately 5% of patients with ulcerative colitis have PSC, though subtler forms of the disease may be present but clinically asymptomatic. PSC is thought to be autoimmune in nature; elevated levels of autoantibodies and an increased expression of human leukocyte antigen (HLA) class II molecules on biliary epithelial cells have been observed.84,85 Approximately two-thirds of patients are male and younger than 45 years of age at the time of diagnosis.86 Patients with PSC may be completely asymptomatic (up to 44%) or have signs of advanced disease at the time of diagnosis.83 Commonly, the diagnosis is made in symptomatic patients after endoscopic retrograde cholangiopancreatography (ERCP) has been performed to evaluate elevated liver enzymes, including alkaline phosphatase and γ-glutamyltransferase. Symptomatic patients have a waxing and waning course and may present with complaints of fatigue (75%), pruritus (25% to 70%), jaundice (30% to 69%), abdominal pain (16% to 37%), and weight loss (10% to 34%).83 Complications secondary to progression to cirrhosis are less common and include ascites, variceal bleeding, and acute cholangitis.
The diagnosis of PSC is suggested by a history of inflammatory bowel disease in the setting of elevated liver enzymes and is established by cholangiography. The disease process may range from a single dominant stricture to, more commonly, diffuse multifocal sclerosis of the intrahepatic and extrahepatic bile ducts. Pathologically, bile ductular proliferation, periductal fibrosis and inflammation, ductopenia, and, less commonly, obliterative fibrous cholangitis may be present. PSC may be considered a premalignant condition. Like ulcerative colitis, PSC confers a significant risk of malignant transformation of the biliary mucosa, which is one of the considerations in considering patients for transplantation.87 Cholangiocarcinoma develops in 1% to 2% of patients per year after the diagnosis of PSC, with an overall risk of up to 10%.88 Screening for premalignant changes with ERCP and cytologic examination and serum testing of CA19-9 have been helpful, though both have limited specificity and sensitivity. Recently, it has been proposed that newer analysis methods of biliary material obtained at screening ERCP, including fluorescence in situ hybridization (FISH), may be used for chromosomal abnormalities associated with cancer with an increase in the sensitivity.89
There is no generally accepted medical therapy to treat PSC, so therapeutic efforts are directed toward complications of the disease. The major complications include biliary obstruction, cirrhosis, and cholangiocarcinoma. Dominant strictures, defined as stenosis less than 1.5 mm in the common bile duct or less than 1 mm in the hepatic duct, are readily treated endoscopically by means of dilation and/or stent placement.90,91 Transplantation should be performed before cancer develops and, generally, the discovery of cancer has been considered an absolute contraindication to transplantation because of poor outcomes. Highly selected patients with localized tumors may be transplanted with an acceptable survival rate with intensive preoperative therapy including radiation and chemotherapy, as proposed by Shaw and implemented on a larger scale by the Mayo group.92
Metabolic and Genetic Disorders. Some of these diseases are listed in Table 59-1. A description of all the metabolic disorders causing liver disease is beyond the scope of this chapter.
In hemochromatosis of the liver, an inborn error of metabolism causes an increased absorption of iron from the gastrointestinal tract. The pathophysiology of iron-induced hepatotoxicity is related to lipid peroxidation induced by iron in periportal regions of the liver. Activation of stellate cells by cytokines released from Kupffer cells that have phagocytosed necrotic hepatocytes injured by iron toxicity is also contributory.93 Over time, the reaction progresses to bridging fibrosis and eventually to a mixed micronodular–macronodular cirrhosis. Treatment includes reduction of iron intake, repeated phlebotomy, and orthotopic liver transplantation.94
Wilson disease is an autosomal, recessively inherited disease caused by a deficiency in hepatocyte transport of copper into the bile. The disease is characterized biochemically by low serum ceruloplasmin levels and clinically by corneal pigmentation (Kayser–Fleischer rings), neuropsychiatric disease, and hepatic cirrhosis.95 As copper accumulates in the liver, periportal inflammation develops that leads to piecemeal and lobular necroses, bridging fibrosis, and a mixed micronodular–macronodular cirrhosis. Treatment options include chelating agents, such as penicillamine, trientine, zinc salts, and orthotopic liver transplantation.
Venous Outflow Obstruction. Cirrhosis may also result from obstruction of the hepatic veins. Causes include chronic right-sided heart failure as a result of severe tricuspid regurgitation, constrictive pericarditis, and the Budd–Chiari syndrome (BCS).
Hepatic dysfunction secondary to passive vascular congestion in the setting of right-sided heart failure and increased right-sided heart pressures is caused by the transmission of increased pressure to the hepatic venous system. This increased pressure leads to sinusoidal congestion, perivenular atrophy, hemorrhagic necrosis, and distortion and enlargement of sinusoidal fenestrations.96 Increased pressure also causes perisinusoidal edema that eventually exceeds the clearance capabilities of hepatic lymphatics, so that ascites develops.97 Grossly, the liver is described as having a “nutmeg” appearance in which areas of hemorrhage are interspersed with relatively normal yellowish parenchyma.97 Histologically, perivenular fibrosis progresses to bridging fibrosis that spares the portal regions. Portal sparing is characteristic of “cardiac cirrhosis.” In addition to causing ascites, chronic vascular congestion can lead to fibrosis in the space of Disse, which compromises nutrient delivery and contributes to portal hypertension and zone 3 hepatocellular injury.98
5 BCS is a rare disease caused by mechanical obstruction of the hepatic veins (Table 59-3). Obstruction may occur at the level of the terminal hepatic veins, the major hepatic veins, or the vena cava and may be caused by obstructing webs or membranes (most commonly in Africa and Asia) or thrombosis secondary to hypercoagulable states and neoplasms (most commonly in the West).
The range of presentations is wide; some patients are completely asymptomatic, whereas acute hepatic failure or cirrhosis develops in others.99 These variations in symptoms are related to the degree and rate of progression of hepatic outflow obstruction. Patients classically present with abdominal pain, hepatomegaly, and ascites. The diagnosis can be made by duplex Doppler ultrasonography, which has a sensitivity of 85% to 95%.100 Identification of collateral vessels to subscapular or intercostal vessels on ultrasonography on ultrasound is a highly sensitive and specific finding.101 Computed tomography (CT) and magnetic resonance imaging (MRI) offer optimal characterization of intrahepatic vascular anatomy and are currently used for the planning of complex interventions.102 Medical management consists of anticoagulation and symptomatic management. Interventional radiologic procedures have now taken the forefront in management of BCS, in which accessible strictures may be treated with angioplasty or stenting. For patients unsuitable for reestablishment of hepatic venous outflow through the above means, transjugular intrahepatic portosystemic shunting (TIPS) is able to decompress both the inferior vena cava and the portal system.101 In patients with fulminant BCS or in those who develop chronic decompensated cirrhosis, liver transplantation becomes an option.
ETIOLOGY
Table 59-3 Etiology

Diagnosis
Although cirrhosis can be asymptomatic for decades, significant information can be obtained by performing a thorough history and physical examination. A history of alcohol abuse, hepatitis, toxin or drug exposure, upper gastrointestinal bleeding, enlarging hemorrhoids, infections, and alteration in mental status suggests the possibility of liver disease. Physical findings associated with cirrhosis are listed in Table 59-4. In addition to these findings, fetor hepaticus, purpura and bruising, decreased body hair, and white nails are common.
Laboratory tests of liver function are indicated if liver disease is suggested by the history and physical examination. Although levels of bilirubin, aspartate aminotransferase, alanine aminotransferase, and alkaline phosphatase are elevated in hepatic disease, the increases are not specific for liver pathology, and levels may be normal even in the setting of significant disease. A very common finding in patients with cirrhosis is thrombocytopenia, caused by hypersplenism and portal hypertension. The platelet growth factor thrombopoietin, which is produced by the liver, has been shown to be decreased in patients with cirrhosis, and this deficit may contribute to the thrombocytopenia associated with hepatic disease.103
The definitive diagnosis of cirrhosis usually requires biopsy, either percutaneous or operative, or gross inspection during laparoscopy or laparotomy. Regardless of the method selected, it is crucial to obtain a large enough specimen to make the diagnosis. Current recommendations suggest that at least 11 portal tracts need to be assessed.104 Several staging systems exist for the grading of fibrosis and cirrhosis, including the International Association for Study of the Liver (IASL), Batts–Ludwig, and Metavir systems. The AASLD recommends the Batts–Ludwig system as it gives a verbal diagnosis rather than numeric categories, and thus may be easier to interpret.104
DIAGNOSIS
Table 59-4 Physical Findings in Cirrhosis

Noninvasive methods to diagnose cirrhosis include ultrasonography, CT, and MRI, which generally reveal an atrophic, nodular liver and an enlarged spleen. More recent work suggests that subtle changes in the hepatic veins may be early markers of cirrhosis.102 Ultrasonographic criteria for cirrhosis include the demonstration of multiple nodular irregularities on the ventral liver surface that are clearly separate from the anterior abdominal wall. Parenchymal texture is altered in the setting of fibrosis, though this feature can be subtle. When these criteria are used, ultrasonography has been shown to have a sensitivity, specificity, and accuracy of approximately 90% in the diagnosis of cirrhosis.105 Ultrasound-based transient elastography, a measure of liver stiffness, is playing an increasingly large role in the noninvasive assessment of fibrosis, with some studies demonstrating a 95% area under the receiver operating characteristic curve (AUROC) for diagnosing grade IV fibrosis (cirrhosis).106
More recently, several laboratory indexes have been found to correlate well with histologic grade of cirrhosis.106,107 The NAFLD fibrosis score, for example, consists of age, BMI, diabetes, platelet count, albumin level, and AST/ALT ratio.108 The AUROC of this scoring system in diagnosing stage 3 or greater fibrosis was 85% in a recent meta-analysis.109 The Fibrotest (LabCorp) is a patented panel of biomarkers consisting of GGT, haptoglobin, bilirubin, apolipoprotein A1, and alpha-2-macroglobulin. The AUROC of Fibrotest for fibrosis greater than or equal to stage 2 has been shown to be up to 87%.110 Indirect evidence of cirrhosis includes endoscopically discovered varices of the esophagus, and the presence of splenomegaly detected by CT or MRI.
Complications
Renal
Renal complications in cirrhosis are intrinsic to functional dysregulation of vascular tone throughout the body.111 Several elements come into play and renal dysfunction is characterized by avid sodium retention despite normovolemia or hypervolemia, dilutional hyponatremia secondary to free water overload, ascites, and ultimately renal failure and the hepatorenal syndrome (HRS). The paradoxical arterial vasoconstriction of the renal arterial bed in the face of global fluid overload is critical to the pathophysiology of HRS.112 Among the complications of cirrhosis, HRS confers the highest risk of mortality.113 Although HRS is the most dramatic renal complication of liver failure, renal dysfunction in patients with advanced liver disease is generally multifactorial.112 Renal insufficiency may develop in a patient with cirrhosis as a direct consequence of the underlying condition (i.e., PBC, amyloidosis), as a consequence of excessive diuretic use in the treatment of ascites and fluid overload, or as a secondary reaction to the release of cytokines or hormones by the liver that alter renal function.
DIAGNOSIS
Table 59-5 Differential Diagnosis of Acute Azotemia in Patients with Liver Disease

Much progress has been made in the understanding of the pathophysiology of HRS focusing on the dysregulation of arterial tone in the end stages of liver disease. Empiric observations that support a functional or “secondary effect” theory include the lack of anatomic renal abnormalities in patients with cirrhosis-related renal dysfunction, the normal function of previously dysfunctional kidneys transplanted into otherwise healthy recipients, and resolution of renal abnormalities after successful hepatic transplantation. Recent clinical studies have defined the critical role of optimizing colloid balance in the patient with cirrhosis114 and the exploration of regulators of renal blood flow such as terlipressin in counteracting the deranged vascular tone.115
Sodium Retention. Patients with cirrhosis who do not have ascites have relatively normal sodium-handling capabilities. Patients in whom ascites develops have a marked inability to excrete sodium. Because of this deficit, sodium intake in excess of renal excretion contributes to fluid overload. Three potential mechanisms have been invoked to explain the cause of sodium retention and the development of ascites in cirrhotic patients. The first is the “underfill” theory, whereby portal hypertension causes an increase in pressure in the splanchnic circulation. Ascites occurs when hepatic lymph production exceeds lymphatic return, with subsequent contraction of the blood volume and renal sodium retention.116 This theory was abandoned when it was shown that patients with cirrhosis were found to have increased, rather than decreased, plasma volume.117 The second is the “overflow” theory, which suggests that the primary defect is inherent to the kidney. Abnormal renal retention of sodium leads to concomitant water retention, expansion of plasma volume, and subsequently edema and ascites.117,118 The third and most currently accepted explanation is the “arterial vasodilation hypothesis,” which suggests that the responsible defect lies within the vascular system, with arterial hypotension as the primary event. Arterial vasodilation in the splanchnic circulation leads to relative peripheral hypovolemia and activation of the renin–angiotensin–aldosterone and sympathetic nervous systems. The effects, in turn, are a release of antidiuretic hormone (arginine vasopressin), enhancement of sodium and water conservation, an increase in effective circulating volume, and edema and ascites.119,120
The cause of the splanchnic vasodilation is unclear. Some evidence suggests that nitric oxide may be the key mediator. Elevations in portal venous nitric oxide have been reported in both animal and human studies,121,122 and inhibitors of nitric oxide production have been shown to reduce the activity of vasoconstrictor systems and enhance renal hemodynamics.122 The exact role of nitric oxide activity in the pathophysiology of renal disease in cirrhosis remains to be determined. Other studies have demonstrated that extracellular fluid volume expansion precedes vasodilation,123 and that pathologic activation of intrahepatic vascular sensors is the key mediator for the ensuing renal salt retention.116
Water Retention. Patients with cirrhosis and ascites may have a marked inability to handle free water. An increased production of antidiuretic hormone, decreased delivery of fluid to the diluting segments of the nephron, and reduced renal production of prostaglandins all may contribute.117 Retention of water leads to dilutional hyponatremia (serum sodium 130 mEq/L), which can cause nausea, vomiting, lethargy, and seizures.124
6 Hepatorenal Syndrome. HRS is a complication of cirrhosis, most often with ascites, characterized by progressive renal failure in the absence of intrinsic renal disease. Renal dysfunction is thought to occur in up to 20% of patients hospitalized with cirrhosis and ascites.125 In a large study of two major Spanish centers, HRS developed in 7.6% of patients admitted for the management of ascites.126 Manifestations of the disease include progressive oliguria, with urine outputs of 400 to 800 mL/d, a rising serum creatinine level, increased cardiac output, and decreased arterial pressure. The disease process is highly variable and is associated with marked renal cortical vasoconstriction induced by activity of the renin–angiotensin–aldosterone and sympathetic nervous systems. In addition, the powerful endothelium-derived vasoconstrictor endothelin-1, combined with decreased renal production of vasodilator prostaglandins, may play a role.
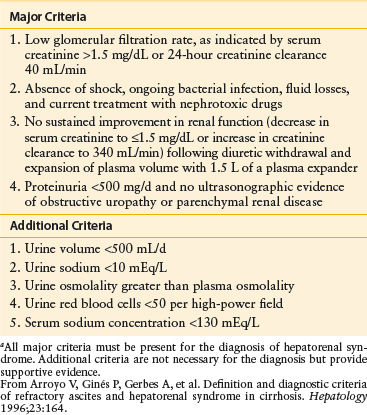
HRS may develop as a result of infection, use of nonsteroidal anti-inflammatory drugs, variceal hemorrhage, or excessive diuretic use in patients who were previously well compensated. The differentiation of HRS from acute renal failure is possible by the laboratory evaluation of urine and serum samples. HRS, however, is virtually indistinguishable by laboratory testing from prerenal azotemia. Both prerenal azotemia and HRS are characterized by extremely low sodium concentrations in the urine, high urine osmolality, high urine-to-plasma ratios of creatinine, and normal urinary sediment (Table 59-5). Criteria for the diagnosis of HRS are listed in Table 59-6.
The treatment of ascites in patients with cirrhosis requires sodium and water restriction in addition to the use of diuretics. Excessive use of these treatment modalities may lead to increases in serum creatinine that can be difficult to distinguish from those of HRS. A failure to respond to cessation of diuretics and fluid challenge suggests HRS. If liver transplantation cannot be performed, patients with HRS usually die within months of the development of severe disease, defined as a serum creatinine level above 2 mg/dL.113
Management of HRS hinges on a three-pronged approach. First is treatment of the triggering event. The most common inciting factor is spontaneous bacterial peritonitis, followed by volume contraction (from multiple etiologies including gastrointestinal bleeding or the overzealous treatment of ascites). Over 70% of cases of acute HRS have been linked to bacterial infections.127 The other two cornerstones of HRS treatment are vasoconstrictors and volume expansion with albumin. The vasopressin-analog terlipressin is commonly used outside the United States. The oral vasopressor midodrine is commonly used in the United States, where terlipressin is not approved by the FDA. Others have examined norepinephrine as a substitute for terlipressin.127 A meta-analysis of all randomized controlled trials of vasopressor use in the treatment of HRS has found a reduction in all-cause mortality when vasopressors are utilized.128 Although advances in medical therapy have made the reversal of HRS possible in some patients, many will ultimately require liver transplantation in order to survive.
Pulmonary
Many pathologic processes in patients with cirrhosis affect pulmonary function. Some reflect an underlying condition that causes both hepatic and pulmonary diseases (i.e., cystic fibrosis, α1-antitrypsin deficiency); others are primary pulmonary processes, such as interstitial lung disease, primary pulmonary hypertension (portopulmonary hypertension [POPH]), and obstructive airway disease. Three main pulmonary manifestations of cirrhosis are discussed here; one is related to increased intra-abdominal pressure secondary to ascites, one is caused by intrapulmonary shunting and is known as the hepatopulmonary syndrome (HPS), and the last is POPH.
The presence of copious ascitic fluid can lead to pulmonary dysfunction by compromising diaphragmatic excursion secondary to increases in intra-abdominal and intrapleural pressures. Ascites may also induce large pleural effusions known as hepatic hydrothorax because of the presence of lymphatic transdiaphragmatic communications between the abdomen and thorax.129 These small diaphragmatic defects are typically found on the right side of the body. Effusions can compress the pulmonary parenchyma and impair gas exchange, so that ventilation–perfusion mismatch and hypoxemia develop. Patients present with worsening pulmonary symptoms in the setting of increasing abdominal girth. Pulmonary function testing reveals decreases in functional residual capacity and total lung capacity.130 Marked improvement in pulmonary function results from medical management of ascites, large-volume paracentesis, and thoracentesis.131,132 These interventions decrease the work of breathing and relieve symptoms. With control of ascites, even in the presence of pleural effusions, no other interventions may be necessary. For patients refractory to the above measures, second- and third-line strategies such as TIPS, indwelling pleural catheter placement, and pleurodesis have been described, although liver transplantation should be pursued in these patients for definitive management.132
HPS occurs in patients with mild to severe hepatic disease and in approximately 10% to 50% of patients with hepatic dysfunction.133,134 Diagnostic criteria for HPS in patients with cirrhosis include PaO2 <80 mm Hg or A–a gradient over 15 mm Hg in patients under 65 years old. The A–a gradient cutoff for patients over 65 is 20 mm Hg.135 Other manifestations include platypnea (increased shortness of breath with movement from a supine to an erect position) and orthodeoxia (decreased oxygen tension on moving from a supine to an erect position). These two positional deficits in pulmonary function are related to the increased number of dilated capillaries in the basal areas of the lung; flow is increased in these vessels while the subject is standing, so that shunting is increased. Physical findings include clubbing and cyanosis of the nail beds and spider angiomata.
Although the underlying cause of hypoxemia in these patients is right-to-left intrapulmonary shunting, ventilation–perfusion mismatch and impaired hypoxic vasoconstriction also play a role. Patients usually present with dyspnea and worsening hypoxemia without evidence of a primary pulmonary process. Initial diagnostic tools include pulse oximetry and arterial blood gas analysis. When significant hypoxemia is found, pulmonary function testing is useful to rule out obstructive or restrictive airway disease. A definitive diagnosis can be obtained by the use of contrast-enhanced echocardiography (bubble study) or technetium-macroaggregated albumin (MAA) scanning, which will confirm the presence of intrapulmonary vascular dilation.136,137 Contrast echocardiography is the most sensitive test, whereby intrapulmonary vascular dilation is demonstrated by a late appearance of microbubbles in the left heart three to six cardiac cycles after injection.138 MAA scanning is complementary to contrast echocardiography in the setting of coexisting intrinsic lung disease with severe hypoxemia, where MAA shunting of >6% suggests HPS as the major contributor to hypoxemia.139 The only effective therapy for this disease is orthotopic liver transplantation, and those with HPS and with a PaO2 <60 mm Hg receive extra priority at listing.138
POPH is defined as a mean pulmonary arterial pressure of greater than 25 mm Hg, pulmonary artery occlusion pressure <15 mm Hg, and pulmonary vascular resistance >240 dyn/s/cm−5 on right heart catheterization in the setting of documented portal hypertension.135 It is a relatively rare (<10%) complication of cirrhosis that, when severe, carries a substantial risk of mortality and is a relative contraindication to liver transplantation.140,141 Screening of patients with cirrhosis by Doppler echocardiography will reveal evidence of elevated right heart pressures, which need to be confirmed by right heart catheterization. Although the high flow state of cirrhosis can elevate right-sided pressures, fixed obstruction of the pulmonary microcirculation is highly lethal and is not likely to improve with liver transplantation. The exact pathophysiology behind POPH is not known, but portal hypertension is a necessary component of the disease process. The hyperdynamic state in portal hypertension may be the main contributory factor, although the severity of POPH does not correlate with the severity of portal hypertension.142 Treatment with prolonged intravenous infusion of prostaglandins has been shown to be effective in improving pulmonary hemodynamics but does not prolong survival.141 The most commonly used drugs in POPH are oral endothelin receptor antagonists such as bosentan and ambrisentan. As stated above, severe POPH (mPAP >50 mm Hg) is an absolute contraindication to liver transplantation. For patients in whom medical therapy can maintain mPAP under 35 mm Hg and PVR under 400 dyn/s/cm−5, priority listing for liver transplantation is available.143
Hepatic Encephalopathy
7 Etiology. Hepatic encephalopathy is a neuropsychiatric syndrome that occurs in the setting of hepatic disease. It is characterized by variable alterations in mental status, ranging from deficits detectable only by detailed psychometric tests to confusion, lethargy, and ultimately frank coma. The disease may present in association with acute hepatic failure, as a consequence of progression of chronic liver disease, or after the creation of a surgical portosystemic shunt. Usually, a precipitating cause, such as an acute variceal hemorrhage or infection, can be found.
The causative agent in hepatic encephalopathy has been the subject of much debate. Most evidence implicates ammonia in the development of this condition. Ammonia is produced during the bacterial digestion of proteins in the gut, is absorbed into the portal circulation, and usually undergoes extensive degradation in the liver.144 Most researchers believe that encephalopathy is caused by products, such as ammonia, derived from the gastrointestinal tract that are usually metabolized by the liver. These agents reach the peripheral circulation as a result of poor hepatic metabolism or through portosystemic shunts that may be physiologic or the result of surgical procedures. In patients with cirrhosis, in addition to the accumulation of ammonia in the blood, the permeability of the brain to ammonia appears to be increased.145 Other suggested etiologic agents for hepatic encephalopathy include γ-aminobutyric acid (GABA), endogenous benzodiazepines,146 branched-chain amino acids (BCAAs) (e.g., tryptophan),147 neurotoxic short-chain fatty acids, mercaptans, phenols,148 and endogenous opiates.149
The following observations suggest that ammonia is the key mediator in hepatic encephalopathy: (a) ammonia levels are increased in 80% to 90% of patients with the condition,150 (b) factors that precipitate hepatic encephalopathy cause increases in ammonia levels, and (c) treatments that relieve hepatic encephalopathy lower ammonia levels.151 Arguments against this hypothesis include the following: (a) levels of ammonia correlate poorly with the severity of hepatic encephalopathy, (b) high ammonia levels alone do not cause encephalopathy, (c) administration of ammonia to patients with cirrhosis but not hepatic encephalopathy does not cause encephalopathy, and (d) treatments that reduce ammonia levels also reduce the levels of other putative toxins.151
Clinical Features. A wide range of neurologic symptoms may occur in patients with hepatic dysfunction. Subtle deficits may include changes in personality, memory loss, alterations in sleep patterns, and minor decreases in intellectual function. Defects may be detectable only by detailed psychometric testing. If no known underlying liver disease is suspected, establishing the cause of an alteration in mental status may be difficult.
TREATMENT
Table 59-7 Treatment of Hepatic Encephalopathy

With progression of disease, asterixis, a rapid repetitive flexion–extension of the wrist that occurs in response to sustained extension of the forearm and fingers, may occur. In addition, stigmata of liver disease are usually evident, including fetor hepaticus and spider angiomas. The combination of asterixis, elevated ammonia levels, and altered mental status in a patient with known liver disease strongly suggests the diagnosis. Electroencephalographic changes are nonspecific and may occur in patients with a variety of other conditions.
Factors that commonly precipitate hepatic encephalopathy include impaired renal function, variceal hemorrhage, constipation, infection, excessive dietary protein, and drugs, especially benzodiazepines and barbiturates.
Treatment. Treatment options for hepatic encephalopathy include correction of the precipitating factors, alterations in diet, bowel cleansing, medications that reduce ammonia production and neutralize its effects, and medications to treat possible neurotransmitter and nutrient deficiencies.
A search for precipitating factors is imperative and includes cultures of urine, sputum, and ascitic fluid; determination of electrolyte abnormalities; screening for viral infection; assessment of overall volume status; drug history; and endoscopy (Table 59-7).
Therapy begins with a trial of volume expansion via intravenous hydration to relieve azotemia and reduce concentrations of toxic substances by dilution. The mainstays of treatment are directed at the removal of nitrogenous compounds from the gut. Most ammonia is produced within the small and large bowels by bacterial metabolism of dietary and endogenous protein. Orally administered cathartics and enemas are the best methods to achieve bowel cleansing. While dietary restriction of protein intake was once a mainstay in the therapy of hepatic encephalopathy, the loss of skeletal muscle, which aids in breakdown of ammonia, is now recognized as an important contributor to hepatic encephalopathy. Current recommendations thus favor regular or increased protein diets along with medical therapy for hepatic encephalopathy. Since patients with cirrhosis often manifest a deficiency of BCAAs compare to aromatic amino acids (which worsen hepatic encephalopathy); supplementation with BCAAs has been shown to improve the rate of recovery from episodic hepatic encephalopathy.152 L-carnitine supplementation has also been demonstrated to improve cognitive deficits and reduce ammonia levels.153
The cathartic of choice is lactulose, a nonabsorbable disaccharide that reaches the distal ileum and colon essentially unmetabolized. Many theories regarding the mechanism of action of lactulose have been proposed. Initially, the presumed mechanism of action was that on reaching the colon, lactulose is metabolized by colonic bacteria to acidic products that lower the pH of the colon. Lowering the pH inhibits the growth of ammonia- and urea-producing bacteria and promotes the growth of Lactobacillus, a bacterium with little proteolytic activity.154 The validity of this theory has been questioned. It appears now that lactulose alters the metabolism of intestinal bacteria by providing carbohydrate, which enhances the bacterial uptake of ammonia.155 Combined with the osmotic diarrhea caused by the cathartic activities of lactulose, this effect leads to an increased excretion of ammonia.
The dosage of lactulose, 45 to 90 g/d, is administered orally, divided into 3 or 4 doses. The dosage can be adjusted to produce two or three soft stools daily. Hourly doses of 30 to 45 mL can be used to induce more rapid improvement during the initial phase of therapy. Symptoms usually abate within 24 hours, but more than 48 hours may be required. Doses can be adjusted if side effects such as flatulence, diarrhea, and electrolyte abnormalities occur.
Nonabsorbable antibiotics have also been used to decrease the number and concentration of ammonia-forming bacteria in the gut. Most experience has accrued for neomycin and metronidazole. These antibiotics are active against gram-negative anaerobes such as Bacteroides, which are considered to be a major source of ammonia production.156,157 The dosage of neomycin, 2 to 8 g/d, is divided into 4 doses and is continued for 4 to 10 days. Multiple double-blinded, randomized trials have determined the efficacy of antibiotics alone or in combination with lactulose. For acute hepatic encephalopathy, studies have shown that neomycin for 4 days is equally as effective as lactulose,157 and metronidazole for 7 days is as effective as neomycin.158 In addition, for chronic hepatic encephalopathy, neomycin for 10 days was equal to lactulose.154
Due to the risk of nephrotoxicity and irreversible ototoxicity, neomycin therapy has fallen out of favor.159 Rifaximin, a minimally absorbed macrolide antibiotic, was approved by the FDA in 2013 for the maintenance therapy of hepatic encephalopathy and is now frequently used in an off-label fashion for acute episodes as well. In a recent randomized controlled trial, the addition of rifaximin to lactulose resulted in a higher proportion of patients experiencing complete reversal of hepatic encephalopathy (76% vs. 50.8%) and decreased mortality (23.1% vs. 49.1%) compared to lactulose and placebo.160 Thus, based on the most current evidence, the treatment of overt hepatic encephalopathy should consist of treatment of precipitating factors, followed by lactulose administration. Rifaximin should be added for patients without improvement in the first 24 hours. Following an episode of overt hepatic encephalopathy, maintenance therapy with lactulose and/or rifaximin should be administered indefinitely for secondary prophylaxis.159
PORTAL HYPERTENSION
8 Portal hypertension is defined as a portal vein pressure above the normal range of 5 to 8 mm Hg. Portal hypertension may also be defined by the hepatic vein–portal vein pressure gradient, which is greater than 5 mm Hg in portal hypertensive states.161 Pressures in the portal venous system are usually measured indirectly via the wedged hepatic venous pressure utilizing a technique similar to that used to determine pulmonary capillary wedge pressure by pulmonary arterial (Swan–Ganz) catheterization.
Anatomy
The venous anatomy of the portal system is relatively constant, with the “usual” anatomy present in 98% of the population (Fig. 59-5). The portal vein is formed by the confluence of the superior mesenteric and splenic veins behind the neck of the pancreas. The inferior mesenteric vein most often joins the splenic vein before the portal vein is formed, but approximately one-third of the time the inferior mesenteric vein joins the superior mesenteric vein. The superior mesenteric vein may not be present, and the portal vein may be formed by multiple small branches from the mesenteric system that join the splenic vein.
Many branches of the portal venous system are affected when portal pressure rises. As pressure increases, blood flow decreases and the pressure in the portal system is transmitted to its branches. This transmission of pressure through branches of the portal system is beneficial in that it decreases overall portal pressure. It also is responsible for many of the complications of portal hypertension, however, because of the resulting dilation of venous tributaries.
The coronary or left gastric vein becomes highly significant in portal hypertension, by diverting portal blood to the veins of the lesser curve of the stomach and the esophagus, leading to the formation of varices. Other important collaterals include the inferior mesenteric vein, which connects with its rectal branches, which, when distended, form hemorrhoids; the umbilical vein in the ligamentum teres of the falciform ligament, which joins the left portal vein and causes abdominal wall veins around the paraumbilical plexus to dilate (caput medusae); the short gastric veins, branches of the splenic vein, which communicate with gastric veins and contribute to gastric varices; and the retroperitoneal veins of Retzius, which communicate with the gastrointestinal veins through the bare areas of the liver where no peritoneal layer separates the abdominal viscera from the retroperitoneum. The retroperitoneal collaterals can also form large splenorenal shunts that may decompress the portal system but are associated with severe encephalopathy.
Physiology
As in any vascular bed, portal hypertension is the product of blood flow and resistance. In nearly all cases, portal hypertension is caused by increased resistance to portal blood flow secondary to cirrhosis, portal vein thrombosis, or hepatic venous obstruction, though in rare instances, an arterioportal fistula may cause flow-related hypertension. Normally, the liver offers little resistance to portal flow because of the porous nature of the hepatic sinusoids and the capacity of the organ to expand. Moreover, the liver has limited control over portal blood flow; it is primarily a passive recipient of splanchnic flow, the primary regulation of which occurs at the level of the splanchnic arterioles.162 As discussed earlier, the deposition of collagen in the space of Disse (capillarization), in addition to the contractile properties of stellate cells, causes an increased resistance to portal blood flow in cirrhosis. In addition, various cytokines and hormones contribute to elevated portal pressures by inducing splanchnic vasodilation and an increase in splanchnic flow.
The increased blood flow through collateral vessels and subsequently increased venous return cause the characteristic hemodynamic features of portal hypertension, which include an increase in cardiac output and total blood volume and a decrease in systemic vascular resistance.163 Arteriovenous shunts within the liver, stomach, and small intestine contribute to the augmented venous return and decreased peripheral vascular resistance. Early in the course of portal hypertension, blood pressure may be normal, but with progression of disease, blood pressure usually falls.164

Figure 59-5. Potential venous collaterals that develop with portal hypertension. The veins of Sappey drain portal blood through the bare areas of the diaphragm and through paraumbilical vein collaterals to the umbilicus. The veins of Retzius form in the retroperitoneum and shunt portal blood from the bowel and other organs to the vena cava.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


