Chronic Heart Failure
KEY CONCEPTS
![]() Heart failure (HF) is a progressive clinical syndrome that can result from any changes in cardiac structure or function that impair the ability of the ventricle to fill with or eject blood. HF may be caused by an abnormality in systolic function, diastolic function, or both. The leading causes of HF are coronary artery disease and hypertension. The primary manifestations of the syndrome are dyspnea, fatigue, and fluid retention.
Heart failure (HF) is a progressive clinical syndrome that can result from any changes in cardiac structure or function that impair the ability of the ventricle to fill with or eject blood. HF may be caused by an abnormality in systolic function, diastolic function, or both. The leading causes of HF are coronary artery disease and hypertension. The primary manifestations of the syndrome are dyspnea, fatigue, and fluid retention.
![]() In response to a decrease in cardiac output, a number of compensatory responses are activated in an attempt to maintain adequate cardiac output, including activation of the sympathetic nervous system (SNS) and the renin–angiotensin–aldosterone system (RAAS), resulting in vasoconstriction and sodium and water retention as well as ventricular hypertrophy/remodeling. These compensatory mechanisms are responsible for the symptoms of HF and contribute to disease progression.
In response to a decrease in cardiac output, a number of compensatory responses are activated in an attempt to maintain adequate cardiac output, including activation of the sympathetic nervous system (SNS) and the renin–angiotensin–aldosterone system (RAAS), resulting in vasoconstriction and sodium and water retention as well as ventricular hypertrophy/remodeling. These compensatory mechanisms are responsible for the symptoms of HF and contribute to disease progression.
![]() Our current understanding of HF pathophysiology is best described by the neurohormonal model. Activation of endogenous neurohormones including norepinephrine, angiotensin II, aldosterone, vasopressin, and numerous proinflammatory cytokines plays an important role in ventricular remodeling and the subsequent progression of HF. Importantly, pharmacotherapy targeted at antagonizing this neurohormonal activation has slowed the progression of HF and improved survival.
Our current understanding of HF pathophysiology is best described by the neurohormonal model. Activation of endogenous neurohormones including norepinephrine, angiotensin II, aldosterone, vasopressin, and numerous proinflammatory cytokines plays an important role in ventricular remodeling and the subsequent progression of HF. Importantly, pharmacotherapy targeted at antagonizing this neurohormonal activation has slowed the progression of HF and improved survival.
![]() Most patients with symptomatic systolic heart failure (SHF) should be routinely treated with an angiotensin-converting enzyme (ACE) inhibitor, a β-blocker, and a diuretic. The benefits of these medications on slowing HF progression, reducing morbidity and mortality, and/or improving symptoms are clearly established. Patients should be treated with a diuretic if there is evidence of fluid retention. Treatment with digoxin may also be considered to improve symptoms and reduce hospitalizations.
Most patients with symptomatic systolic heart failure (SHF) should be routinely treated with an angiotensin-converting enzyme (ACE) inhibitor, a β-blocker, and a diuretic. The benefits of these medications on slowing HF progression, reducing morbidity and mortality, and/or improving symptoms are clearly established. Patients should be treated with a diuretic if there is evidence of fluid retention. Treatment with digoxin may also be considered to improve symptoms and reduce hospitalizations.
![]() In patients with SHF, ACE inhibitors improve survival, slow disease progression, reduce hospitalizations, and improve quality of life. The doses for these agents should be targeted at those shown in clinical trials to improve survival. When ACE inhibitors are contraindicated or not tolerated, an angiotensin II receptor blocker or the combination of hydralazine and isosorbide dinitrate is a reasonable alternative. Patients with asymptomatic left ventricular dysfunction and/or a previous myocardial infarction (MI) (Stage B of the American College of Cardiology [ACC]/American Heart Association [AHA] classification scheme) should also receive ACE inhibitors, with the goal of preventing symptomatic HF and reducing mortality.
In patients with SHF, ACE inhibitors improve survival, slow disease progression, reduce hospitalizations, and improve quality of life. The doses for these agents should be targeted at those shown in clinical trials to improve survival. When ACE inhibitors are contraindicated or not tolerated, an angiotensin II receptor blocker or the combination of hydralazine and isosorbide dinitrate is a reasonable alternative. Patients with asymptomatic left ventricular dysfunction and/or a previous myocardial infarction (MI) (Stage B of the American College of Cardiology [ACC]/American Heart Association [AHA] classification scheme) should also receive ACE inhibitors, with the goal of preventing symptomatic HF and reducing mortality.
![]() The β-blockers carvedilol, metoprolol succinate, and bisoprolol have been shown to prolong survival, decrease hospitalizations and need for transplantation, and cause “reverse remodeling” of the left ventricle. These agents are recommended for all patients with a reduced left ventricular ejection fraction. Therapy must be instituted at low doses, with slow upward titration to the target dose.
The β-blockers carvedilol, metoprolol succinate, and bisoprolol have been shown to prolong survival, decrease hospitalizations and need for transplantation, and cause “reverse remodeling” of the left ventricle. These agents are recommended for all patients with a reduced left ventricular ejection fraction. Therapy must be instituted at low doses, with slow upward titration to the target dose.
![]() Although chronic diuretic therapy frequently is used in HF patients, it is not mandatory. Diuretic therapy along with sodium restriction is required only in those patients with peripheral edema and/or pulmonary congestion. Many patients will need continued diuretic therapy to maintain euvolemia after fluid overload is resolved.
Although chronic diuretic therapy frequently is used in HF patients, it is not mandatory. Diuretic therapy along with sodium restriction is required only in those patients with peripheral edema and/or pulmonary congestion. Many patients will need continued diuretic therapy to maintain euvolemia after fluid overload is resolved.
![]() Digoxin does not improve survival in patients with SHF but does provide symptomatic benefits. Digoxin doses should be adjusted to achieve plasma concentrations of 0.5 to 1 ng/mL (0.6 to 1.3 nmol/L); higher plasma concentrations are not associated with additional benefits but may be associated with increased risk of toxicity.
Digoxin does not improve survival in patients with SHF but does provide symptomatic benefits. Digoxin doses should be adjusted to achieve plasma concentrations of 0.5 to 1 ng/mL (0.6 to 1.3 nmol/L); higher plasma concentrations are not associated with additional benefits but may be associated with increased risk of toxicity.
![]() Aldosterone antagonists, also known as mineralocorticoid receptor antagonists, reduce mortality in patients with SHF and New York Heart Association (NYHA) class II to IV symptoms and thus should be strongly considered in these patients provided that potassium and renal function can be carefully monitored. Aldosterone antagonists should also be considered soon after MI in patients with left ventricular dysfunction and either HF or diabetes.
Aldosterone antagonists, also known as mineralocorticoid receptor antagonists, reduce mortality in patients with SHF and New York Heart Association (NYHA) class II to IV symptoms and thus should be strongly considered in these patients provided that potassium and renal function can be carefully monitored. Aldosterone antagonists should also be considered soon after MI in patients with left ventricular dysfunction and either HF or diabetes.
![]() The combination of hydralazine and nitrates improves the composite end point of mortality, hospitalizations for HF, and quality of life in African Americans receiving standard therapy. Current guidelines recommend the addition of hydralazine and nitrates to self-described African Americans with moderate to severe symptoms despite therapy with ACE inhibitors, diuretics, and β-blockers. This combination is also reasonable to consider in all patients who continue to have symptoms despite optimized therapy with an ACE inhibitor (or angiotensin receptor blocker [ARB]) and β-blocker. Hydralazine and a nitrate might be reasonable in patients unable to tolerate either an ACE inhibitor or ARB because of renal insufficiency, hyperkalemia, or possibly hypotension.
The combination of hydralazine and nitrates improves the composite end point of mortality, hospitalizations for HF, and quality of life in African Americans receiving standard therapy. Current guidelines recommend the addition of hydralazine and nitrates to self-described African Americans with moderate to severe symptoms despite therapy with ACE inhibitors, diuretics, and β-blockers. This combination is also reasonable to consider in all patients who continue to have symptoms despite optimized therapy with an ACE inhibitor (or angiotensin receptor blocker [ARB]) and β-blocker. Hydralazine and a nitrate might be reasonable in patients unable to tolerate either an ACE inhibitor or ARB because of renal insufficiency, hyperkalemia, or possibly hypotension.
![]() Treatment of HF with a preserved ejection fraction should be targeted at symptom reduction, causal clinical disease, and underlying basic mechanisms. Patients with HF and a preserved EF may be treated differently than those with systolic dysfunction.
Treatment of HF with a preserved ejection fraction should be targeted at symptom reduction, causal clinical disease, and underlying basic mechanisms. Patients with HF and a preserved EF may be treated differently than those with systolic dysfunction.
INTRODUCTION
![]()
![]() Heart failure (HF) is a progressive clinical syndrome that can result from any abnormality in cardiac structure or function that impairs the ability of the ventricle to fill with or eject blood.1 HF may be caused by an abnormality in systolic function, diastolic function, or both. Making the distinction is important because the prevalence, prognosis, and treatment of HF may be quite different depending on whether the predominant mechanism causing the symptoms is systolic or diastolic dysfunction. HF is the final common pathway for numerous cardiac disorders including those affecting the pericardium, heart valves, and myocardium. Diseases that adversely affect ventricular diastole (filling), ventricular systole (contraction), or both can lead to HF. For many years it was believed that reduced myocardial contractility, or systolic dysfunction (i.e., reduced left ventricular ejection fraction [LVEF]), was the sole disturbance in cardiac function responsible for HF. However, it is now recognized that large numbers of patients with the HF syndrome have relatively normal systolic function (i.e., normal LVEF). This is now referred to as HF with preserved LVEF (HFpEF) and is believed to be primarily due to diastolic dysfunction of the heart.1 Recent estimates suggest approximately 50% of patients with HF have preserved LVEF with disturbances in relaxation (lusitropic) properties of the heart, or diastolic dysfunction.2 However, regardless of the etiology of HF, the underlying pathophysiologic process and principal clinical manifestations (fatigue, dyspnea, and often volume overload) are similar and appear to be independent of the initial cause. Historically, this disorder was commonly referred to as congestive HF; the preferred nomenclature is now HF since a patient may have the clinical syndrome of HF without having symptoms of congestion. This chapter will focus on treatment of patients with chronic HF from reduced as well as preserved LVEF. Chapter 5 will discuss the treatment of acute decompensated HF.2
Heart failure (HF) is a progressive clinical syndrome that can result from any abnormality in cardiac structure or function that impairs the ability of the ventricle to fill with or eject blood.1 HF may be caused by an abnormality in systolic function, diastolic function, or both. Making the distinction is important because the prevalence, prognosis, and treatment of HF may be quite different depending on whether the predominant mechanism causing the symptoms is systolic or diastolic dysfunction. HF is the final common pathway for numerous cardiac disorders including those affecting the pericardium, heart valves, and myocardium. Diseases that adversely affect ventricular diastole (filling), ventricular systole (contraction), or both can lead to HF. For many years it was believed that reduced myocardial contractility, or systolic dysfunction (i.e., reduced left ventricular ejection fraction [LVEF]), was the sole disturbance in cardiac function responsible for HF. However, it is now recognized that large numbers of patients with the HF syndrome have relatively normal systolic function (i.e., normal LVEF). This is now referred to as HF with preserved LVEF (HFpEF) and is believed to be primarily due to diastolic dysfunction of the heart.1 Recent estimates suggest approximately 50% of patients with HF have preserved LVEF with disturbances in relaxation (lusitropic) properties of the heart, or diastolic dysfunction.2 However, regardless of the etiology of HF, the underlying pathophysiologic process and principal clinical manifestations (fatigue, dyspnea, and often volume overload) are similar and appear to be independent of the initial cause. Historically, this disorder was commonly referred to as congestive HF; the preferred nomenclature is now HF since a patient may have the clinical syndrome of HF without having symptoms of congestion. This chapter will focus on treatment of patients with chronic HF from reduced as well as preserved LVEF. Chapter 5 will discuss the treatment of acute decompensated HF.2
EPIDEMIOLOGY
HF is an epidemic public health problem in the United States.3 Nearly 6 million Americans have HF with an additional 670,000 cases diagnosed each year.3 Unlike most other cardiovascular diseases, the incidence and prevalence of HF are increasing and are expected to continue to increase over the next few decades as the population ages. A large majority of patients with HF are elderly, with multiple comorbid conditions that influence morbidity and mortality.3 Improved survival after myocardial infarction (MI) is thought to be a likely contributor to the increased incidence and prevalence of HF.4 Annual hospital discharges for HF now total over 1 million, although recent data suggest hospitalization rates are declining in the Medicare population.4 However, HF remains the most common hospital discharge diagnosis in individuals over age 65.3 The disorder also has a tremendous economic impact, with this expected to increase markedly as the baby-boom generation ages. Current estimates suggest annual expenditures for HF of approximately $39 billion, with the majority of these costs spent on hospitalized patients.5 Thus, HF is a major medical problem, with substantial economic impact that is expected to become even more significant as the population ages.
Despite prodigious advances in our understanding of the etiology, pathophysiology, and pharmacotherapy of HF, the prognosis for patients with this disorder remains grim. Although the mortality rates have declined over the last 50 years, the overall 5-year survival remains approximately 50% for all patients with a diagnosis of HF, with mortality increasing with symptom severity.3,4,6 Death is classified as sudden in about 40% of patients, implicating serious ventricular arrhythmias as the underlying cause.1 Factors affecting the prognosis of patients with HF include, but are not limited to, age, gender, LVEF, renal function, natriuretic peptide plasma concentrations, diabetes, extent of underlying coronary artery disease, blood pressure (BP), HF etiology, and drug or device therapy. Recent models incorporating these and other factors enable clinicians to develop reliable estimates of an individual patient’s prognosis.5,6
ETIOLOGY
![]()
![]() HF can result from any disorder that affects the ability of the heart to contract (systolic function) and/or relax (diastolic dysfunction); common causes of HF are shown in Table 4-1.7 HF with reduced systolic function (i.e., reduced LVEF) is the classic, more familiar form of the disorder, but current estimates suggest up to 50% of patients with HF have preserved left ventricular systolic function with presumed diastolic dysfunction.2,8 In contrast to systolic heart failure (SHF) that is usually caused by previous MI, patients with preserved LVEF typically are elderly, female, and obese, and have hypertension (HTN), atrial fibrillation, or diabetes.8 Recent data indicate that survival is similar in patients with impaired or preserved LVEF.8
HF can result from any disorder that affects the ability of the heart to contract (systolic function) and/or relax (diastolic dysfunction); common causes of HF are shown in Table 4-1.7 HF with reduced systolic function (i.e., reduced LVEF) is the classic, more familiar form of the disorder, but current estimates suggest up to 50% of patients with HF have preserved left ventricular systolic function with presumed diastolic dysfunction.2,8 In contrast to systolic heart failure (SHF) that is usually caused by previous MI, patients with preserved LVEF typically are elderly, female, and obese, and have hypertension (HTN), atrial fibrillation, or diabetes.8 Recent data indicate that survival is similar in patients with impaired or preserved LVEF.8
TABLE 4-1 Causes of Heart Failure

![]() Coronary artery disease is the most common cause of SHF, accounting for nearly 70% of cases.8,9 MI leads to reduction in muscle mass due to death of affected myocardial cells. The degree to which contractility is impaired depends on the size of the infarction. To attempt to maintain cardiac output (CO), the surviving myocardium undergoes a compensatory remodeling, thus beginning the maladaptive process that initiates the HF syndrome and leads to further injury to the heart. This is discussed in greater detail in Pathophysiology below. Myocardial ischemia and infarction also affect the diastolic properties of the heart by increasing ventricular stiffness and slowing ventricular relaxation. Thus, MI frequently results in systolic and diastolic dysfunction.
Coronary artery disease is the most common cause of SHF, accounting for nearly 70% of cases.8,9 MI leads to reduction in muscle mass due to death of affected myocardial cells. The degree to which contractility is impaired depends on the size of the infarction. To attempt to maintain cardiac output (CO), the surviving myocardium undergoes a compensatory remodeling, thus beginning the maladaptive process that initiates the HF syndrome and leads to further injury to the heart. This is discussed in greater detail in Pathophysiology below. Myocardial ischemia and infarction also affect the diastolic properties of the heart by increasing ventricular stiffness and slowing ventricular relaxation. Thus, MI frequently results in systolic and diastolic dysfunction.
Impaired systolic function is a cardinal feature of dilated cardiomyopathies. Although the cause of reduced contractility frequently is unknown, abnormalities such as interstitial fibrosis, cellular infiltrates, cellular hypertrophy, and myocardial cell degeneration are seen commonly on histologic examination. Inherited forms of dilated as well as hypertrophic cardiomyopathies may also occur.9,10
Pressure or volume overload causes ventricular hypertrophy, which attempts to return contractility to a near-normal state. If the pressure or volume overload persists, the remodeling process results in alterations in the geometry of the hypertrophied myocardial cells and is accompanied by increased collagen deposition in the extracellular matrix. Thus, both systolic and diastolic functions may be impaired.7 Examples of pressure overload include systemic or pulmonary HTN and aortic or pulmonic valve stenosis.
HTN remains an important cause and/or contributor to HF in many patients, particularly women, the elderly, and African Americans.1,8 The role of HTN should not be underestimated because HTN is an important risk factor for ischemic heart disease and thus is also present in a high percentage of the patients with this disorder. HF is a largely preventable disorder such that appropriate management of lifestyle risk factors (e.g., HTN, coronary heart disease, smoking, obesity, physical activity, diabetes, etc.) is key to minimize the risk of HF development.
PATHOPHYSIOLOGY
Normal Cardiac Function
To understand the pathophysiologic processes in HF, a basic understanding of normal cardiac function is necessary. CO is defined as the volume of blood ejected per unit time (L/min) and is the product of heart rate (HR) and stroke volume (SV):
CO = HR × SV
The relationship between CO and mean arterial pressure (MAP) is:
MAP = CO × systemic vascular resistance (SVR)
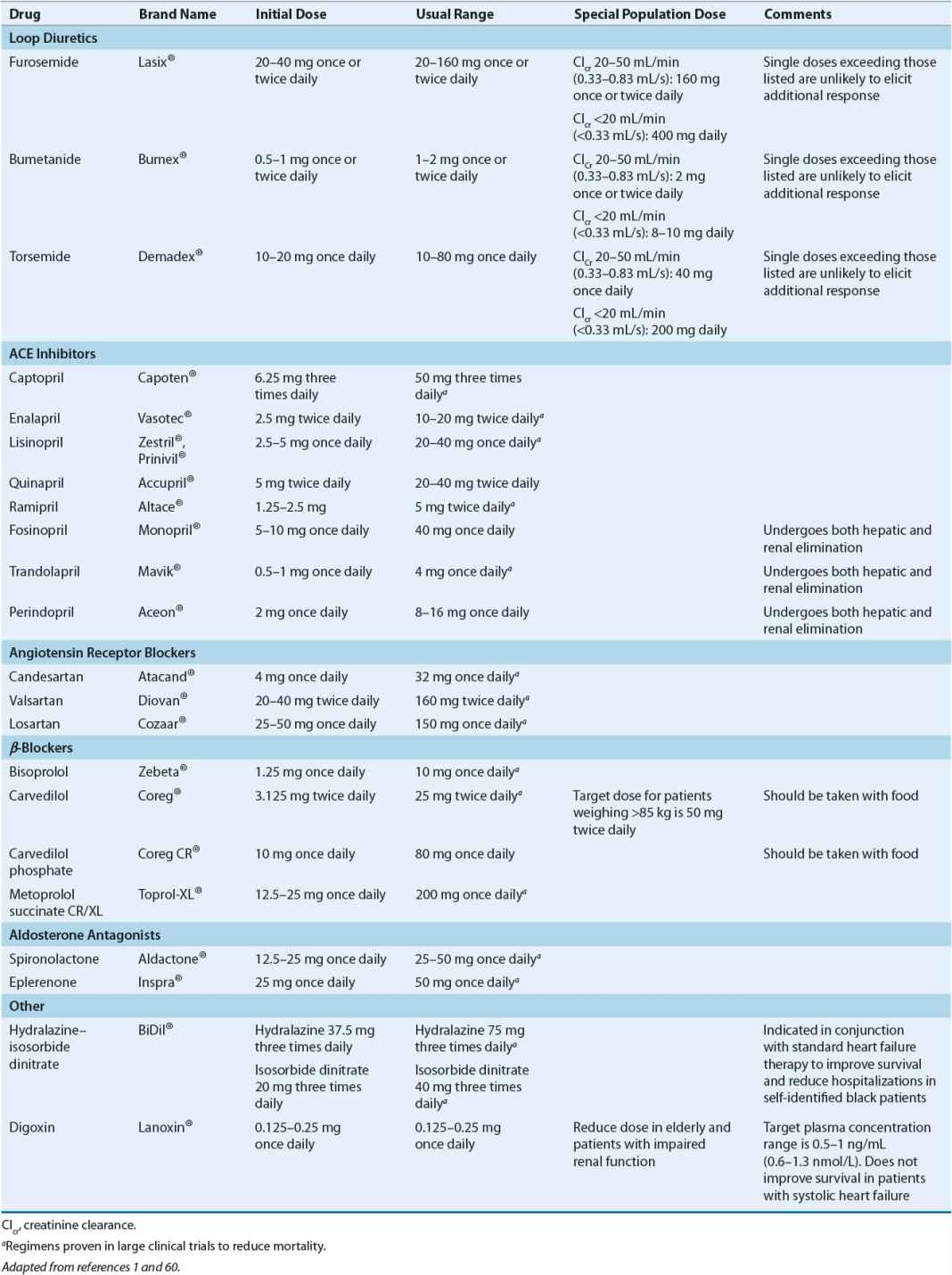
HR is controlled by the autonomic nervous system. SV, or the volume of blood ejected during systole, depends on preload, afterload, and contractility.7 As defined by the Frank-Starling mechanism, the ability of the heart to alter the force of contraction depends on changes in preload. As myocardial sarcomere length is stretched, the number of cross-bridges between thick and thin myofilaments increases, resulting in an increase in the force of contraction. The length of the sarcomere is determined primarily by the volume of blood in the ventricle; therefore, left ventricular end-diastolic volume (LVEDV) is the primary determinant of preload. In normal hearts, the preload response is the primary compensatory mechanism such that a small increase in end-diastolic volume results in a large increase in CO. Because of the relationship between pressure and volume in the heart, left ventricular end-diastolic pressure (LVEDP) is often used in the clinical setting to estimate preload. The hemodynamic measurement used to clinically estimate LVEDP is the pulmonary capillary wedge pressure (PCWP), also known as the pulmonary artery occlusion pressure (PAOP). Afterload is a more complex physiologic concept that can be viewed pragmatically as the sum of forces preventing active forward ejection of blood by the ventricle. Major components of afterload are ejection impedance, wall tension, and regional wall geometry. In patients with left ventricular systolic dysfunction, an inverse relationship exists between afterload (estimated clinically by SVR) and SV such that increasing afterload causes a decrease in SV (Fig. 4-1). Contractility is the intrinsic property of cardiac muscle describing fiber shortening and tension development.
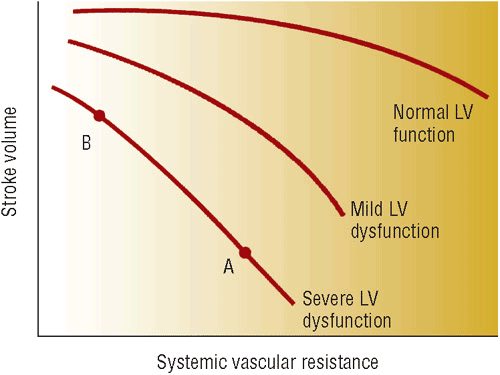
FIGURE 4-1 Relationship between stroke volume and systemic vascular resistance. In an individual with normal left ventricular (LV) function, increasing systemic vascular resistance has little effect on stroke volume. As the extent of LV dysfunction increases, the negative, inverse relationship between stroke volume and systemic vascular resistance becomes more important (B to A).
HF with a preserved ejection fraction can be defined as a condition in which myocardial relaxation and filling are impaired and incomplete. The ventricle is unable to accept an adequate volume of blood from the venous system, does not fill at low pressure, and/or is unable to maintain normal SV. In its most severe form, HFpEF results in overt symptoms of HF. In modest HFpEF, symptoms of dyspnea and fatigue occur only during stress or activity, when HR and end-diastolic volume increase. In its mildest form, HFpEF can be manifested as a slow or delayed pattern of relaxation and filling with little or no elevation in diastolic pressure and few or no cardiac symptoms. The congestive symptoms that occur with HFpEF are a manifestation of increased pulmonary venous pressures. HFpEF is caused by impaired myocardial relaxation and/or increased diastolic stiffness. When HF is caused by a predominant abnormality in diastolic function, the ventricular chamber is not enlarged, and EF may be normal or even elevated.2,11 Figure 4-2 shows the pressure–volume relationship in a patient with normal versus abnormal diastolic function. Changes in the myocardium are associated with a shift upward and to the left of the pressure–volume curve, so that for any increase in LV volume, diastolic pressure rises to a much greater level than normally would occur. Clinically, patients present with reduced exercise tolerance and dyspnea when they have elevated LV diastolic pressures. Patients with HFpEF have a predominant abnormality in diastolic function, whereas patients with SHF have a predominant abnormality in systolic function of the LV.
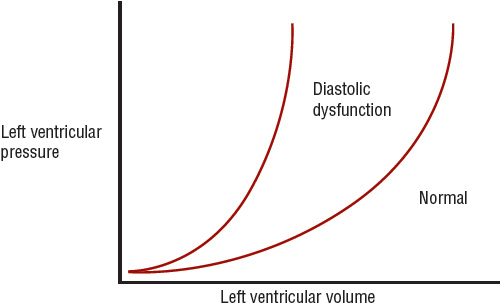
FIGURE 4-2 Diastolic pressure–volume relationship in a normal patient (right trace) and a patient with diastolic dysfunction (left trace).
Recent data suggest that HFpEF may also be associated with abnormalities in endothelial and ventricular reserve function. During physical exertion, CO increases through integrated enhancements in venous return, contractility, HR, and peripheral vasodilation. The vasodilation that normally occurs during exercise is impaired in HFpEF. Pulmonary HTN is also a common finding. Abnormalities in each of these components of normal exercise reserve function have been identified in HFpEF and all may contribute to pathophysiology in individual patients.2
Compensatory Mechanisms in Heart Failure
![]() HF is a progressive disorder initiated by any event that impairs the ability of the heart to contract and/or relax resulting in a decrease in CO. The index event may have an acute onset, as with MI, or the onset may be slow, as with long-standing HTN. Regardless of the index event, the decrease in CO results in activation of compensatory responses to maintain the circulation.7,10,12 These compensatory responses include: (a) tachycardia and increased contractility through sympathetic nervous system (SNS) activation, (b) the Frank-Starling mechanism, whereby an increase in preload results in an increase in SV, (c) vasoconstriction, and (d) ventricular hypertrophy and remodeling. Compensatory responses evolved to provide short-term support to maintain circulatory homeostasis after acute reductions in BP or renal perfusion. However, the persistent decline in CO in HF results in long-term activation of these compensatory responses resulting in the complex functional, structural, biochemical, and molecular changes important for the development and progression of HF. The beneficial and detrimental effects of these compensatory responses are described below and are summarized in Table 4-2.
HF is a progressive disorder initiated by any event that impairs the ability of the heart to contract and/or relax resulting in a decrease in CO. The index event may have an acute onset, as with MI, or the onset may be slow, as with long-standing HTN. Regardless of the index event, the decrease in CO results in activation of compensatory responses to maintain the circulation.7,10,12 These compensatory responses include: (a) tachycardia and increased contractility through sympathetic nervous system (SNS) activation, (b) the Frank-Starling mechanism, whereby an increase in preload results in an increase in SV, (c) vasoconstriction, and (d) ventricular hypertrophy and remodeling. Compensatory responses evolved to provide short-term support to maintain circulatory homeostasis after acute reductions in BP or renal perfusion. However, the persistent decline in CO in HF results in long-term activation of these compensatory responses resulting in the complex functional, structural, biochemical, and molecular changes important for the development and progression of HF. The beneficial and detrimental effects of these compensatory responses are described below and are summarized in Table 4-2.
TABLE 4-2 Beneficial and Detrimental Effects of the Compensatory Responses in Heart Failure

Tachycardia and Increased Contractility
The increase in HR and contractility that rapidly occurs in response to a drop in CO is primarily due to release of norepinephrine (NE) from adrenergic nerve terminals, although parasympathetic nervous system activity is also diminished.12 Loss of atrial contribution to ventricular filling also can occur (atrial fibrillation, ventricular tachycardia), reducing ventricular performance even more. Because ionized calcium is sequestered into the sarcoplasmic reticulum and pumped out of the cell during diastole, the shortened diastolic time with increases in HR also results in a higher average intracellular calcium concentration during diastole, increasing actin–myosin interaction, augmenting the active resistance to fibril stretch, and reducing lusitropy. Conversely, the higher average calcium concentration translates into greater filament interaction during systole, generating more tension.7 Increasing HR also increases myocardial oxygen demand. If ischemia is induced or worsened, both diastolic and systolic functions may become impaired, and SV can drop precipitously. In addition, polymorphisms in genes coding for adrenergic receptors (e.g., β1– and α2c-receptors) and their signaling pathways may affect the risk for development of HF and alter the response to endogenous NE.13
Fluid Retention and Increased Preload
Augmentation of preload is another compensatory response that is rapidly activated in response to decreased CO. Renal perfusion in HF is reduced due to both depressed CO and redistribution of blood away from nonvital organs. The kidney interprets the reduced perfusion as an ineffective blood volume, resulting in activation of the renin–angiotensin–aldosterone system (RAAS) in an attempt to maintain BP and increase renal sodium and water retention. Reduced renal perfusion and increased sympathetic tone also stimulate renin release from juxtaglomerular cells in the kidney. As shown in Figure 4-3, renin is responsible for conversion of angiotensinogen to angiotensin I. Angiotensin I is converted to angiotensin II by angiotensin-converting enzyme (ACE). Angiotensin II may also be generated via non–ACE-dependent pathways. Angiotensin II stimulates aldosterone release from the adrenal gland, thereby providing an additional mechanism for renal sodium and water retention. As intravascular volume increases secondary to sodium and water retention, left ventricular volume and pressure (preload) increase, sarcomeres are stretched, and the force of contraction is enhanced.7 While the preload response is the primary compensatory mechanism in normal hearts, the chronically failing heart usually has exhausted its preload reserve.7 As shown in Figure 4-4, increases in preload will increase SV only to a certain point. Once the flat portion of the curve is reached, further increases in preload will only lead to pulmonary or systemic congestion, a detrimental result.7 Figure 4-4 also shows that the curve is flatter in patients with left ventricular dysfunction. Consequently, a given increase in preload in a patient with HF will produce a smaller increment in SV than in an individual with normal ventricular function.
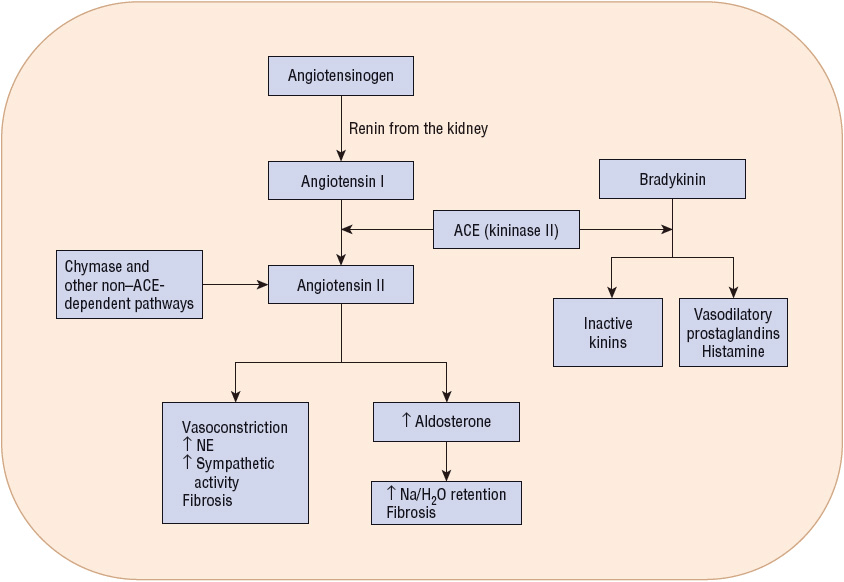
FIGURE 4-3 Physiology of the renin–angiotensin–aldosterone system. Renin produces angiotensin I from angiotensinogen. Angiotensin I is cleaved to angiotensin II by angiotensin-converting enzyme (ACE). Angiotensin II has a number of physiologic actions that are detrimental in heart failure. Note that angiotensin II can be produced in a number of tissues, including the heart, independent of ACE activity. ACE is also responsible for the breakdown of bradykinin. Inhibition of ACE results in accumulation of bradykinin that, in turn, enhances the production of vasodilatory prostaglandins.
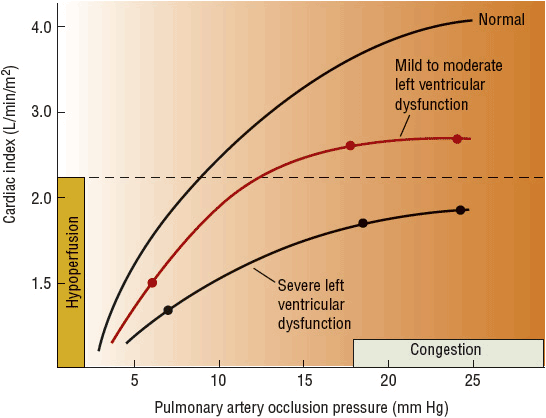
FIGURE 4-4 Relationship between cardiac output (shown as cardiac index which is CO/BSA) and preload (shown as pulmonary artery occlusion pressure).
Vasoconstriction and Increased Afterload
Vasoconstriction occurs in patients with HF to help redistribute blood flow away from nonessential organs to coronary and cerebral circulations to support BP, which may be reduced secondary to a decrease in CO (MAP = CO × SVR).7 A number of neurohormones likely contribute to the vasoconstriction, including NE, angiotensin II, endothelin-1 (ET-1), neuropeptide Y, urotensin II, and arginine vasopressin (AVP).7 Vasoconstriction impedes forward ejection of blood from the ventricle, further depressing CO and heightening the compensatory responses. The failing ventricle is exquisitely sensitive to changes in afterload (Fig. 4-1). Thus, increases in afterload often potentiate a vicious cycle of continued worsening and downward spiraling of the HF state.
Ventricular Hypertrophy and Remodeling
![]() While the signs and symptoms of HF are closely associated with the items described above, the progression of HF appears to be independent of the patient’s hemodynamic status. It is now recognized that left ventricular hypertrophy and remodeling are key components in the pathogenesis of progressive myocardial failure.7 Ventricular hypertrophy is a term used to describe an increase in ventricular muscle mass. Cardiac or ventricular remodeling is a broader term describing changes in both myocardial cells and extracellular matrix that result in changes in the size, shape, structure, and function of the heart. These progressive changes in ventricular structure and function ultimately result in a change in shape of the left ventricle from an ellipse to a sphere. This change in ventricular size and shape serves to further depress the mechanical performance of the heart, increases regurgitant flow through the mitral valve, and, in turn, fuels the continued progression of remodeling. Ventricular hypertrophy and remodeling can occur in association with any condition that causes myocardial injury. The onset of the remodeling process precedes the development of HF symptoms.
While the signs and symptoms of HF are closely associated with the items described above, the progression of HF appears to be independent of the patient’s hemodynamic status. It is now recognized that left ventricular hypertrophy and remodeling are key components in the pathogenesis of progressive myocardial failure.7 Ventricular hypertrophy is a term used to describe an increase in ventricular muscle mass. Cardiac or ventricular remodeling is a broader term describing changes in both myocardial cells and extracellular matrix that result in changes in the size, shape, structure, and function of the heart. These progressive changes in ventricular structure and function ultimately result in a change in shape of the left ventricle from an ellipse to a sphere. This change in ventricular size and shape serves to further depress the mechanical performance of the heart, increases regurgitant flow through the mitral valve, and, in turn, fuels the continued progression of remodeling. Ventricular hypertrophy and remodeling can occur in association with any condition that causes myocardial injury. The onset of the remodeling process precedes the development of HF symptoms.
Cardiac remodeling is a complex process that affects the heart at the molecular and cellular levels. Key elements in the process are shown in Figure 4-5. Collectively, these events result in progressive changes in myocardial structure and function such as cardiac hypertrophy, myocyte loss, and alterations in the extracellular matrix. The progression of the remodeling process leads to reductions in myocardial systolic and/or diastolic function that, in turn, results in further myocardial injury, perpetuating the remodeling process and the decline in left ventricular performance. Angiotensin II, NE, ET, aldosterone, vasopressin, and numerous inflammatory cytokines, as well as substances under investigation, that are activated both systemically and locally in the heart and vasculature play an important role in initiating the signal transduction cascade responsible for ventricular remodeling. Although these mediators produce harmful effects on the heart, their increased circulating and tissue concentrations are also toxic to other organs and serve as an important reminder that HF is a systemic as well as a cardiac disorder.12
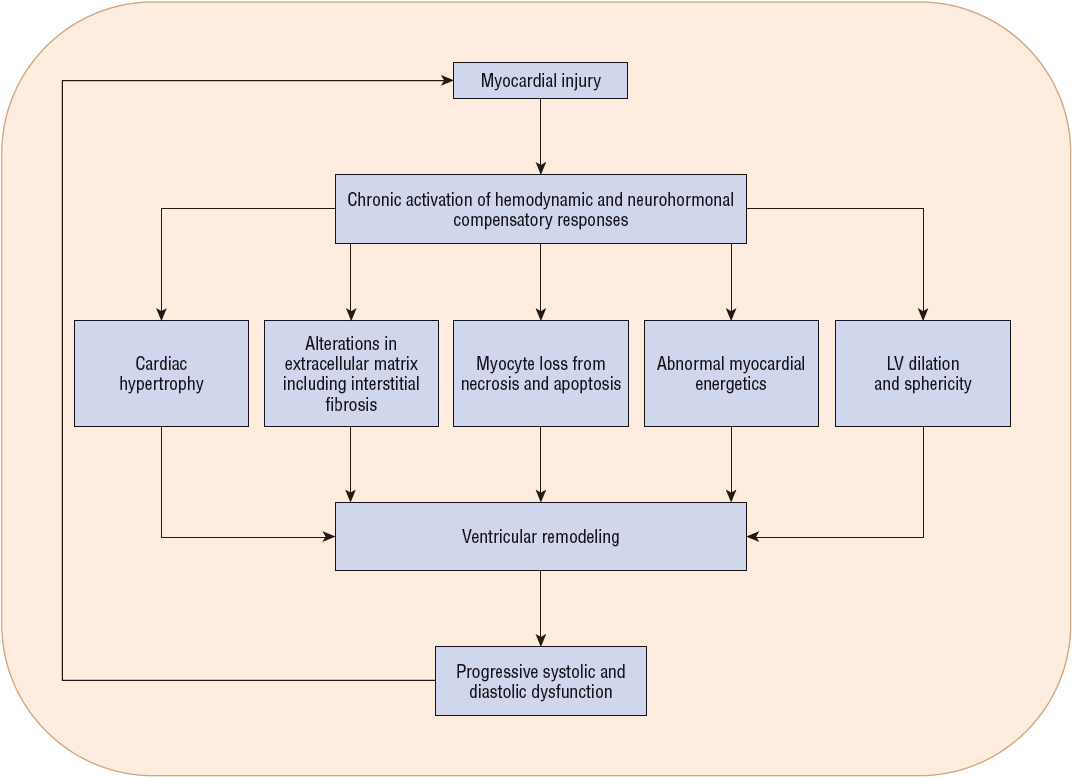
FIGURE 4-5 Key components of the pathophysiology of cardiac remodeling. Myocardial injury (e.g., myocardial infarction) results in the activation of a number of hemodynamic and neurohormonal compensatory responses in an attempt to maintain circulatory homeostasis. Chronic activation of the neurohormonal systems results in a cascade of events that affect the myocardium at the molecular and cellular levels. These events lead to the changes in ventricular size, shape, structure, and function known as ventricular remodeling. The alterations in ventricular function result in further deterioration in cardiac systolic and diastolic functions that further promotes the remodeling process.
Pressure overload (and probably hormonal activation) associated with HTN produces a concentric hypertrophy (increase in the ventricular wall thickness without chamber enlargement). Conversely, eccentric left ventricular hypertrophy (myocyte lengthening with increased chamber size with minimal increase in wall thickness) characterizes the hypertrophy seen in patients with systolic dysfunction or previous MI. As the myocytes undergo change, so do various components of the extracellular matrix. For example, collagen degradation may lead to myocyte slippage, fibroblast proliferation, and increased fibrillar collagen synthesis, resulting in fibrosis and stiffening of the entire myocardium. Thus, a number of important ventricular changes that occur with remodeling include alterations in the geometry of the heart from an elliptical to a spherical shape, increases in ventricular mass (from myocyte hypertrophy), and changes in ventricular composition (especially the extracellular matrix) and volumes, all of which contribute to the impaired cardiac function. If the event producing cardiac injury is acute (e.g., MI), the ventricular remodeling process begins immediately. However, it is the progressive nature of this process that results in continual worsening of the HF state, and thus is now the major focus for identification of therapeutic targets. In fact, HF pharmacotherapy associated with decreased mortality, and/or slowing the progression of the disease, produces these effects largely by slowing or reversing ventricular remodeling, a process often referred to as reverse remodeling.
The Neurohormonal Model of Heart Failure and Therapeutic Insights It Provides
![]()
![]() Over the years, several different paradigms have guided our understanding of the pathophysiology and treatment of HF.7 The early paradigm is often called the cardiorenal model, where the problem was viewed as excess sodium and water retention, and diuretic therapy was the main therapeutic approach. Next, the cardiocirculatory model focused on impaired CO (viewed as being due to both reduced pumping capacity of the heart and systemic vasoconstriction). Treatment strategies here focused on positive inotropes and, later, vasodilators, as the primary therapies to overcome reduced CO. While the therapeutic approaches associated with these paradigms provided some symptomatic benefits, they did little to slow progression of the disease. In fact, the detrimental effects of positive inotropic drugs on survival highlighted the inadequacy of the cardiocirculatory model to explain the progressive nature of HF.
Over the years, several different paradigms have guided our understanding of the pathophysiology and treatment of HF.7 The early paradigm is often called the cardiorenal model, where the problem was viewed as excess sodium and water retention, and diuretic therapy was the main therapeutic approach. Next, the cardiocirculatory model focused on impaired CO (viewed as being due to both reduced pumping capacity of the heart and systemic vasoconstriction). Treatment strategies here focused on positive inotropes and, later, vasodilators, as the primary therapies to overcome reduced CO. While the therapeutic approaches associated with these paradigms provided some symptomatic benefits, they did little to slow progression of the disease. In fact, the detrimental effects of positive inotropic drugs on survival highlighted the inadequacy of the cardiocirculatory model to explain the progressive nature of HF.
Balanced (arterial and venous) vasodilation with ACE inhibitors was the basis for initial clinical trials with these agents. Subsequent discovery that ACE inhibitors provided benefits beyond their vasodilating effects, followed by the positive results with β-adrenergic receptor blockers and aldosterone antagonists, led to the current paradigm used to describe HF pathogenesis: the neurohormonal model.7,14 This model recognizes an initiating event (e.g., MI, long-standing HTN) that leads to decreased CO and begins the “HF state.” The problem then moves beyond the heart, and it becomes a systemic disease whose progression is mediated largely by neurohormones and autocrine/paracrine factors that drive myocyte injury, oxidative stress, inflammation, and extracellular matrix remodeling. While the former paradigms still guide us to some extent in the symptomatic management of the disease (e.g., diuretics and digoxin), it is the latter paradigm that helps us understand disease progression and, more important, the ways to slow disease progression. In the sections that follow, key neurohormones and autocrine/paracrine factors, sometimes now collectively termed biomarkers, are described with respect to their role in HF and its progression. The benefits of current and investigational drug therapies can be better understood through a solid understanding of the neurohormones they regulate/affect. Although the neurohormonal model provides a logical framework for our current understanding of HF progression and the role of various medications in attenuating this progression, it must be emphasized that this model does not completely explain HF progression. For example, drug therapies that target the neurohormonal perturbations in HF usually only slow the progressive nature of the disorder rather than completely stop it. Ongoing research will likely identify additional targets for drug therapy.
Angiotensin II
Of the neurohormones and autocrine/paracrine factors that play an important role in SHF pathophysiology, angiotensin II is probably the best understood.7,15 Although circulating angiotensin II produced from ACE activity is the most familiar route for generation of angiotensin II, recent evidence indicates that this hormone is synthesized directly in the myocardium through non–ACE-dependent pathways that also contribute to HF pathophysiology.
Angiotensin II has multiple actions that contribute to its detrimental effects in HF. It is a potent vasoconstrictor mediated by binding to the angiotensin type 1 (AT1) receptor in the vasculature and it also causes release of AVP and ET-1. Angiotensin II facilitates release of NE from adrenergic nerve terminals, heightening SNS activation. It promotes sodium retention through direct effects on the renal tubules and by stimulating aldosterone release. Its vasoconstriction of the efferent glomerular arteriole helps to maintain renal perfusion pressure in patients with severe HF or impaired renal function. Finally, angiotensin II, and many of the neurohormones released in response to angiotensin II, plays a central role in stimulating ventricular hypertrophy, remodeling, myocyte apoptosis, oxidative stress, inflammation, and alterations in the myocardial extracellular matrix. Clinical data suggest that attenuating angiotensin II–mediated effects contributes substantially to the benefits of ACE inhibitor–treated and angiotensin receptor blocker (ARB)–treated patients with SHF.15,16 The favorable effects of ACE inhibitors and ARBs on hemodynamics, symptoms, hospitalizations, quality of life, and survival highlight the importance of angiotensin II in the pathophysiology of this disorder.
Norepinephrine
As described earlier in this chapter, NE plays a central role in the tachycardia, vasoconstriction, and increased contractility and plasma renin activity in HF.12 Plasma NE concentrations are elevated in correlation with the degree of HF, and patients with the highest plasma NE concentrations have the poorest prognosis.7,17 Excessive SNS activation causes downregulation of β1-receptors, with a subsequent loss of sensitivity to receptor stimulation. Excess catecholamines increase the risk of arrhythmias and can cause myocardial cell loss by stimulating both necrosis and apoptosis. Finally, NE contributes to ventricular hypertrophy and remodeling. The detrimental effects of SNS activation are further highlighted by the clinical trials of chronic therapy with β-agonists, phosphodiesterase inhibitors, or other drugs that cause SNS activation, since these agents are uniformly associated with increased mortality. Conversely, β-blockers, ACE inhibitors, and digoxin all help to decrease SNS activation, through various mechanisms, and are beneficial in HF. Thus, it is clear that NE plays a critical role in the pathophysiology of the HF state.
Aldosterone
Aldosterone-mediated sodium retention and its key role in volume overload and edema have long been recognized as important components of the HF syndrome.18 Circulating aldosterone is increased in HF due to stimulation of its synthesis and release from the adrenal cortex by angiotensin II and due to decreased hepatic clearance from reduced hepatic perfusion. Recent studies demonstrate direct effects of aldosterone on the heart that may be even more important than sodium retention in HF pathophysiology. Chief among these is the ability of aldosterone to produce interstitial cardiac fibrosis through increased collagen deposition in the extracellular matrix of the heart. By increasing the stiffness of the myocardium, cardiac fibrosis may decrease systolic function and impair diastolic function. Current research shows that extra-adrenal production of aldosterone in the heart, kidneys, and vascular smooth muscle also contributes to the progressive nature of HF through target organ fibrosis and vascular remodeling. Induction of a systemic proinflammatory state, increased oxidative stress, wasting of soft tissues and bone, secondary hyperparathyroidism, and mineral/micronutrient dyshomeostasis are other important pathologic actions of aldosterone that directly contribute to ventricular remodeling and HF progression.19 Aldosterone also may increase the risk of ventricular arrhythmias through a number of mechanisms, including creation of reentrant circuits as a result of fibrosis, inhibition of cardiac NE reuptake, depletion of intracellular potassium and magnesium, and impairment of parasympathetic traffic. Other detrimental effects of aldosterone include insulin resistance and endothelial and baroreceptor dysfunction. Clinical trials with the aldosterone antagonists spironolactone20 and eplerenone21,22 showing significant reductions in morbidity and mortality in patients with HF provide compelling evidence of the important role of aldosterone in the initiation and progression of this syndrome.
Natriuretic Peptides
The natriuretic peptide family has three members, atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP), and C-type natriuretic peptide (CNP).22 Of these, BNP is the one that is most useful in the diagnosis and management of HF.23,24 BNP is synthesized and released from the ventricle in response to pressure or volume overload. BNP plasma concentrations are elevated in patients with HF functioning to increase natriuresis and diuresis and attenuate activation of the RAAS and SNS.
The development of easily performed commercial assays for BNP and the related biologically inactive peptide, NT-proBNP, resulted in widespread interest in the role of these peptides as a biomarker for prognostic, diagnostic, and therapeutic use. In patients with chronic HF, the degree of elevation in BNP levels is closely associated with increased mortality, risk of sudden death, symptoms, and hospital readmission.23,24 Accurate diagnosis of acute decompensated HF in acute care settings is often difficult since many of the symptoms (e.g., dyspnea) mimic those of other disorders such as pulmonary disease or obesity. The most well-established clinical application of BNP testing is in the urgent care setting where the BNP or NT-proBNP assay is useful when combined with clinical evaluation for differentiating dyspnea secondary to either SHF or HFpEF from other causes. The Breathing Not Properly study evaluated 452 patients with echocardiography within 30 days of an emergency department visit. Of the 452 patients, 165 (36.5%) had EF >45% (mean EF 59%).25 In the patients with preserved EF who had been admitted to the hospital for dyspnea, BNP levels were significantly lower than those found in patients with SHF (413 pg/mL vs. 821 pg/mL [119 pmol/L vs. 237 pmol/L]). However, there was considerable overlap in the BNP levels in patients with HFpEF compared with those in patients without HF, making BNP levels less useful. Furthermore, the sensitivity, specificity, and predictive accuracy of BNP levels in HFpEF are limited in part because BNP is altered by age, adiposity, gender, and other factors. Similar findings have been documented with NT-proBNP. In a study of 68 symptomatic patients with isolated HFpEF (EF >50%), NT-proBNP was significantly increased in patients with isolated HFpEF and correlated with disease severity. Compared with conventional echocardiography, Doppler imaging, and heart catheterization, NT-proBNP exhibited the best negative predictive value for detection of HFpEF.26
Much interest has focused on the benefits of serial measurement of BNP as a target to guide drug therapy, primarily diuretics. Recent studies evaluating this approach have not shown consistent improvement in long-term outcomes compared with standard medical therapy.23,24 As a result, current guidelines do not support the routine use of serial measurement of BNP in the management of chronic HF.1,27
Arginine Vasopressin
AVP is a pituitary peptide hormone that regulates renal water excretion and plasma osmolality.7,14 Plasma concentrations of AVP are elevated in patients with HF, supporting its role in the pathophysiology of this disorder. The physiologic effects of AVP are mediated through the V1a, V1b, and V2 receptors. Stimulation of these receptors by increased circulating AVP results in several maladaptive responses including: (a) increased renal free water reabsorption in the face of plasma hypoosmolality resulting in volume overload and hyponatremia; (b) increased arterial vasoconstriction that contributes to reduced CO; and (c) stimulation of remodeling by cardiac hypertrophy and extracellular matrix collagen deposition.
Given the importance of AVP in HF, recent efforts have focused on the development of AVP antagonist drugs for treatment of acute and chronic HF. By blocking the AVP receptor, these agents primarily increase free water excretion (i.e., an “aquaretic” effect). Although clinical trials with tolvaptan demonstrate improvements in acute symptoms and increases in serum sodium and urine output without affecting HR, BP, renal function, or other electrolytes, no improvements in morbidity and mortality were seen.28
Other Circulating Biomarkers
The role of other biomarkers in HF pathogenesis, risk stratification, and identification of patients at risk for developing HF, and as potential therapeutic targets is an area of extensive ongoing research.13,14,23 Many of these biomarkers (e.g., ET, C-reactive protein and other inflammatory cytokines, copeptin, procalcitonin) are involved in inflammation, oxidative stress, extracellular matrix remodeling, myocyte injury and stress, and kidney injury that drive the systemic response to the failing left ventricle. It is hoped that data from investigations will lead to improved understanding of disease pathophysiology, prognosis, and targets for therapy.
Factors Precipitating/Exacerbating Heart Failure
Although significant advancements have been made in treatment, symptom exacerbation, to the point that hospitalization is required, is a common and growing problem in patients with chronic HF. Hospitalization for HF exacerbation consumes large amounts of healthcare dollars and significantly impairs the patient’s quality of life; thus, there is great interest in identifying and then remedying factors that increase the risk of decompensation. Appropriate therapy can often maintain patients in a “compensated” state, indicating that they are relatively symptom-free. However, there are many aggravating or precipitating factors that may cause a previously compensated patient to develop worsened symptoms necessitating hospitalization. Often, these precipitating factors are reversible or treatable, such that a thorough evaluation for their presence is imperative.
Cardiac events are a frequent cause of worsening HF.1,29 Myocardial ischemia and infarction are potentially reversible causes that must be carefully considered since nearly 70% of HF patients have coronary artery disease. Revascularization should be considered in appropriate patients. Atrial fibrillation occurs in up to 10% to 50% of patients with HF and is associated with increased morbidity and mortality.30,31 It can exacerbate HF through rapid ventricular response and loss of atrial contribution to ventricular filling. Conversely, HF can precipitate atrial fibrillation by increasing atrial distension from ventricular volume overload. Control of ventricular response, maintenance of sinus rhythm in appropriate patients, and prevention of thromboembolism are important elements in the treatment of HF patients with atrial fibrillation. Uncontrolled HTN is also an important contributing factor and should be treated according to current guidelines.30,31
Noncardiac events are also associated with HF decompensation. Pulmonary infections frequently cause worsening HF. Many of these events would be preventable with more widespread use of the pneumococcal and influenza vaccines. Pulmonary embolus, diabetes, worsening renal function, hypothyroidism, and hyperthyroidism should also be considered.
Nonadherence with prescribed HF medications or with dietary recommendations (e.g., sodium intake and fluid restriction) is also a common cause of HF exacerbation.29,32 Recent estimates indicate that nonadherence is an important contributor to poor outcomes and that socioeconomically disadvantaged patients appear to be disproportionately affected.
A number of drugs can precipitate or exacerbate HF by one or more of the following mechanisms: (a) negative inotropic effects; (b) direct cardiotoxicity; or (c) increased sodium and/or water retention (Table 4-3).33 The resulting symptoms are typically those associated with volume overload, but in more severe cases hypoperfusion may also be present. Nonsteroidal antiinflammatory drugs (NSAIDs) are increasingly recognized for their ability to exacerbate HF and increase risk of hospitalization and mortality through volume retention, decreased renal function, and increased BP.33
TABLE 4-3 Drugs That May Precipitate or Exacerbate Heart Failure

What should be evident is that many of the precipitating factors are preventable, particularly through appropriate healthcare professional intervention. Specifically, patient education and counseling by a pharmacist should be able to identify and address inadequate HF therapy, identify medication nonadherence, and administration of drugs or the presence of drug–drug interactions that may worsen HF (Table 4-3). A careful medication history is an important aspect of evaluating the cause(s) of HF exacerbation. Discontinuation of medications known to exacerbate HF may help prevent hospitalizations. Use of medications such as antiarrhythmic agents, particularly disopyramide, dronedarone, and flecainide, and nondihydropyridine calcium channel blockers are important precipitants of exacerbations. The widespread use of NSAIDs, particularly the nonprescription ones that many patients perceive as having a low risk of adverse effects, is also problematic and should be discouraged. The thiazolidinedione (TZD) hypoglycemic drugs, rosiglitazone and pioglitazone, cause fluid retention and weight gain that may exacerbate HF. Current guidelines indicate these agents should not be used in patients with New York Heart Association (NYHA) class III or IV HF.1 Thus, many of the factors precipitating HF exacerbations (nonadherence, inadequate/inappropriate drug therapy, uncontrolled HTN, etc.) are amenable to pharmacist intervention. Thus, the value of the pharmacist’s role in careful and repeated education of patients and monitoring of the drug regimen should not be underestimated.34,35 Attention to these factors may make an important contribution to reducing the risk of hospitalization and improving the patient’s quality of life.
CLINICAL PRESENTATION
Signs and Symptoms
![]() The primary manifestations of HF are dyspnea and fatigue, which lead to exercise intolerance, and fluid overload, which can result in peripheral edema and pulmonary congestion.1,33,36 The presence of these signs and symptoms may vary considerably from patient to patient such that some patients have dyspnea but no signs of fluid retention, whereas others may have marked volume overload with few complaints of dyspnea or fatigue. However, many patients have both dyspnea and volume overload. Clinicians should remember that symptom severity often does not correlate with the degree of LV dysfunction. Patients with a low LVEF (less than 20% to 25%) may be asymptomatic, whereas those with preserved LVEF may have significant symptoms. It is also important to note that symptoms can vary considerably over time in a given patient, even in the absence of changes in ventricular function or medications.
The primary manifestations of HF are dyspnea and fatigue, which lead to exercise intolerance, and fluid overload, which can result in peripheral edema and pulmonary congestion.1,33,36 The presence of these signs and symptoms may vary considerably from patient to patient such that some patients have dyspnea but no signs of fluid retention, whereas others may have marked volume overload with few complaints of dyspnea or fatigue. However, many patients have both dyspnea and volume overload. Clinicians should remember that symptom severity often does not correlate with the degree of LV dysfunction. Patients with a low LVEF (less than 20% to 25%) may be asymptomatic, whereas those with preserved LVEF may have significant symptoms. It is also important to note that symptoms can vary considerably over time in a given patient, even in the absence of changes in ventricular function or medications.
Pulmonary congestion arises as the left ventricle fails and is unable to accept and eject the increased blood volume that is delivered to it or if pulmonary pressures are elevated due to a stiff, nondistensible ventricle. Consequently, pulmonary venous and capillary pressures rise, leading to interstitial and bronchial edema, increased airway resistance, and dyspnea. The associated signs and symptoms may include (a) dyspnea (with or without exertion), (b) orthopnea, (c) paroxysmal nocturnal dyspnea (PND), and (d) pulmonary edema. Exertional dyspnea occurs when there is a reduction in the level of exertion that causes breathlessness. This is typically described as more breathlessness than was associated previously with a specific activity (e.g., vacuuming, stair climbing). As HF progresses, many patients eventually have dyspnea at rest.
Orthopnea is dyspnea that occurs with assumption of the supine position. It occurs within minutes of recumbency and is due to reduced pooling of blood in the lower extremities and abdomen. Orthopnea is relieved almost immediately by sitting upright and typically is prevented by elevating the head with pillows. An increase in the number of pillows required to prevent orthopnea (e.g., a change from “two-pillow” to “three-pillow” orthopnea) suggests worsening HF. Attacks of PND typically occur after 2 to 4 hours of sleep; the patient awakens from sleep with a sense of suffocation. The attacks are due to severe pulmonary and bronchial congestion, leading to shortness of breath, cough, and wheezing. The reasons these attacks occur at night are unclear but may include (a) reduced pooling of blood in the lower extremities and abdomen (as with orthopnea), (b) slow resorption of interstitial fluid from sites of dependent edema, (c) normal reduction in sympathetic activity that occurs with sleep (e.g., less support for the failing ventricle), and (d) normal depression in respiratory drive that occurs with sleep.
Rales (crackling sounds heard on auscultation) are present in the lung bases due to transudation of fluid into alveoli. The rales typically are bibasilar, but if heard unilaterally, they are usually right-sided. Rales are not present in most patients with chronic HF even though there is volume overload. This is thought to be due to a compensatory increase in lymphatic drainage. Detection of rales is usually indicative of a rapid onset of worsening HF rather than the amount of excess fluid volume. A third heart sound, or S3 gallop, is heard frequently in patients with left ventricular failure and may be due to elevated atrial pressure and altered distensibility of the ventricle.
Pulmonary edema is the most severe form of pulmonary congestion, and is caused by accumulation of fluid in the interstitial spaces and alveoli. In HF patients, it is the result of increased pulmonary venous pressure. The patient experiences extreme breathlessness and anxiety and may cough pink, frothy sputum. Pulmonary edema can be terrifying for the patient, causing a feeling of suffocation or drowning. Patients with pulmonary edema may also report any of the above-mentioned signs or symptoms of pulmonary congestion.
Systemic congestion is associated with a number of signs and symptoms. Jugular venous distension (JVD) is the simplest and most reliable sign of fluid overload. Examination of the right internal jugular vein with the patient at a 45° angle is the preferred method for assessing JVD. The presence of JVD more than 4 cm above the sternal angle suggests systemic venous congestion. In patients with mild systemic congestion, JVD may be absent at rest, but application of pressure to the abdomen will cause an elevation of JVD (hepatojugular reflux).
Peripheral edema is a cardinal finding in HF. Edema usually occurs in dependent parts of the body, and thus is seen as ankle or pedal edema in ambulatory patients, although it may be manifested as sacral edema in bedridden patients. Adults typically have a 10-lb (4.5-kg) fluid weight gain before trace peripheral edema is evident; therefore, patients with acute decompensated HF may have no clinical evidence of systemic congestion except weight gain. Body weight is thus the best short-term end point for evaluating fluid status. Nonfluid weight gain and loss of muscle mass due to cardiac cachexia are potential confounders for long-term use of weight as a marker for fluid status. Hepatomegaly and ascites are other signs of systemic congestion.
Patients with HF may exhibit signs and symptoms of low CO alone or in addition to volume overload. The primary complaint associated with hypoperfusion is fatigue. Poor appetite or early satiety may be due to limited perfusion of the GI tract. Conversely, patients with such GI complaints may simply be experiencing gut edema. Subjective measures of low CO include worsening renal function, cool extremities, altered mental status, resting tachycardia, and narrow pulse pressure.
Diagnosis
No single test is available to confirm the diagnosis of HF—it is a clinical syndrome associated with specific signs and symptoms.1,27,36 Because HF can be caused or worsened by multiple cardiac and noncardiac disorders, many of which may be treatable or reversible, accurate diagnosis is essential for development of therapeutic strategies. HF is often initially suspected in a patient based on his or her symptoms. However, signs and symptoms lack sensitivity for diagnosing HF since they are frequently found with many other disorders. Even in patients with known HF, there is poor correlation between the presence or severity of symptoms and the hemodynamic abnormality. With few exceptions, HFpEF cannot be distinguished from SHF on the basis of the history, physical examination, chest x-ray, and ECG alone. The frequency with which patients have symptoms and signs of HF on physical examination or chest x-ray is not dependent on whether they have SHF or HFpEF.37 Patients with HFpEF are often elderly, hypertensive women.37
CLINICAL PRESENTATION Heart Failure
A complete history and physical examination targeted at identifying cardiac or noncardiac disorders or behaviors that may cause or hasten HF development or progression are essential in the initial patient evaluation. However, the physical examination cannot distinguish between HF due to decreased systolic function and that due to preserved systolic function. A careful medication history should also be obtained with a focus on use of ethanol, tobacco, illicit drugs (e.g., cocaine or methamphetamine), vitamins and supplements (including herbal or “natural” supplements), NSAIDs, and antineoplastic agents (anthracyclines, cyclophosphamide, trastuzumab, imatinib).
Particular attention should be paid to cardiovascular risk factors and to other disorders that can cause or exacerbate HF such as HTN, diabetes, atrial fibrillation, dyslipidemia, tobacco use, sleep-disordered breathing, and thyroid disease. Since coronary artery disease is the cause of HF in many patients, evaluation of the possibility of coronary disease is essential, especially in men. If coronary artery disease is detected, appropriate revascularization procedures may then be considered. The patient’s volume status should be documented by assessing the body weight, JVD, and presence or absence of pulmonary congestion and peripheral edema. Laboratory testing may assist in identification of disorders that cause or worsen HF. The initial evaluation should include a complete blood count, serum electrolytes (including calcium and magnesium), assessment of renal and hepatic function, urinalysis, lipid profile, hemoglobin A1C, thyroid function tests, chest x-ray, and 12-lead ECG. There are no specific ECG abnormalities associated with HF, but findings may help detect coronary artery disease or conduction abnormalities that could affect prognosis and guide treatment decisions. Measurement of BNP or NT-proBNP may also assist in differentiating dyspnea caused by HF from other causes.
Although the history, physical examination, and laboratory tests provide important insight into the underlying cause of HF, the echocardiogram is the single most useful test in the evaluation of the patient. The echocardiogram is used to assess abnormalities in cardiac structure and function and should include evaluation of the pericardium, myocardium, and heart valves, and quantification of the LVEF to determine if systolic or diastolic dysfunction is present.
TREATMENT OF CHRONIC HEART FAILURE
Desired Outcomes
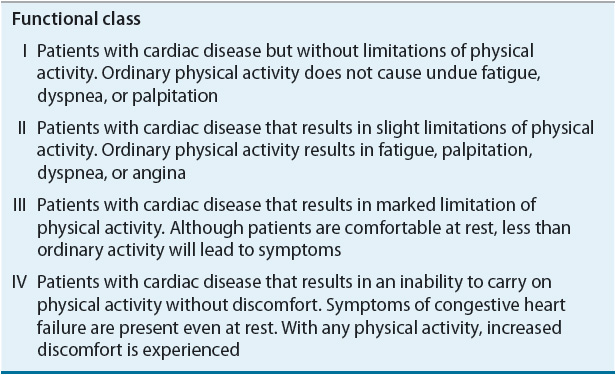
The goals of therapy in management of chronic HF are to improve the patient’s quality of life, relieve or reduce symptoms, prevent or minimize hospitalizations for exacerbations of HF, slow progression of the disease process, and prolong survival. Pharmacotherapy plays a key role in achieving these goals.1 In addition, identification of risk factors for HF development and recognition of its progressive nature have led to increased emphasis on preventing the development of this disorder. With this in mind, the American College of Cardiology (ACC)/American Heart Association (AHA) guidelines for the evaluation and management of chronic HF utilize a staging system that not only recognizes the evolution and progression of the disorder but also emphasizes risk factor modification and preventive treatment strategies (Fig. 4-6).1 The four stages of this system differ from the NYHA functional classification (Table 4-4) with which most clinicians are familiar. The NYHA system is primarily intended to classify symptoms according to the clinician’s subjective evaluation and does not recognize preventive measures or the progression of the disorder. A patient’s symptoms can change frequently over a short period of time due to changes in medications, diet, intercurrent illnesses, etc. For example, a patient with ACC/AHA Stage C HF with NYHA class IV symptoms such as marked volume overload could rapidly improve to class I to II with aggressive diuretic therapy. Despite these limitations, this system can be useful for monitoring patients and is widely used in HF studies. In contrast, and consistent with the progressive nature of HF, a patient’s ACC/AHA HF stage could not improve (e.g., go from Stage C to Stage B) even though the patient’s symptoms could fluctuate from NYHA class IV to I. In addition, the ACC/AHA staging system provides a more comprehensive framework for evaluation, prevention, and treatment of HF.
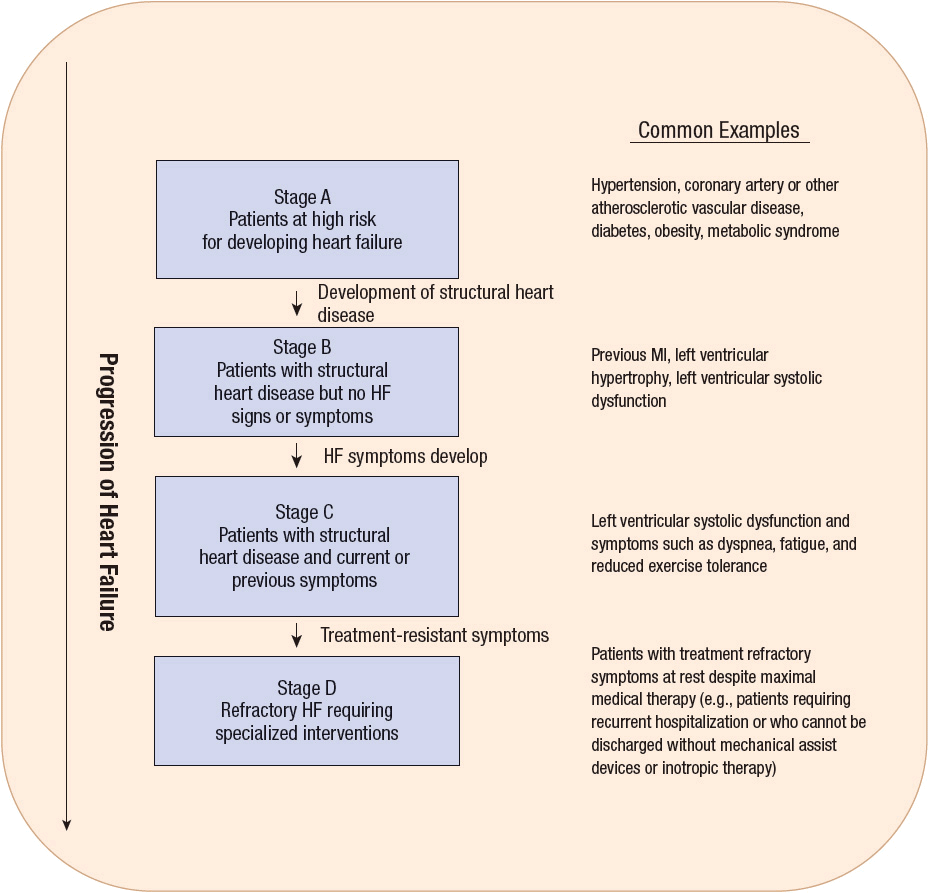
FIGURE 4-6 ACC/AHA heart failure staging system. (Adapted with permission from Hunt SA, Abraham WT, Chin MH, et al. Circulation 2009; 119:e391–e479.)
TABLE 4-4 New York Heart Association Functional Classification
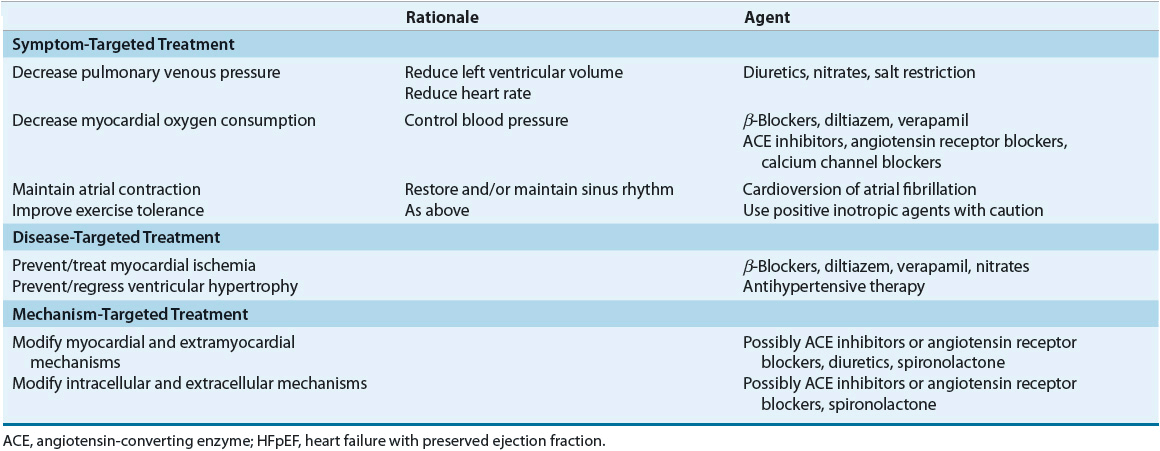
The general principles used to guide the treatment of SHF are based on numerous large, randomized, double-blind, multicenter trials. Until recently, no such randomized trials had been performed in patients with HFpEF. Consequently, the guidelines for the management of HFpEF are based primarily on clinical investigations in relatively small groups of patients, clinical experience, and concepts based on the knowledge and understanding of the pathophysiology of the disease process. The treatment regimen outlined in Table 4-5 applies to patients with HFpEF who have clear manifestations of congestion either at rest or with exertion. Whether treatment of asymptomatic diastolic dysfunction confers any benefit has not been demonstrated.
TABLE 4-5 Targeted Approach to Treatment of HFpEF
General Measures
The complexity of the HF syndrome necessitates a comprehensive approach to management that includes accurate diagnosis, identification and treatment of risk factors, elimination or minimization of precipitating factors, appropriate pharmacologic and nonpharmacologic therapy, and close monitoring and followup.
The first step in management of chronic HF is to determine the etiology (see Table 4-1) and/or any precipitating factors. Appropriate treatment of underlying disorders (e.g., hyperthyroidism, valvular heart disease) may obviate the need for specific HF treatment. Revascularization or antiischemic therapy in patients with coronary disease may reduce HF symptoms. Drugs that aggravate HF (see Table 4-3) should be discontinued if possible.
Restriction of physical activity reduces cardiac workload and is recommended for virtually all patients with acute congestive symptoms. However, once the patient’s symptoms have stabilized and excess fluid is removed, restrictions on physical activity are discouraged.38 Exercise training may improve quality of life and yield trends toward reduced hospitalizations and death from cardiovascular causes.34,38 Current guidelines support exercise training programs in stable patients to improve clinical status.1
Because a major compensatory response in HF is sodium and water retention, restriction of dietary sodium and fluid intake is an important lifestyle intervention. Mild (<3 g/day) to moderate (<2 g/day) sodium restriction, in conjunction with daily measurement of weight, should be implemented to minimize volume retention and allow use of lower and safer diuretic doses. The typical American diet contains 8 to 10 g of sodium per day, so most patients would need to reduce their intake by over 50%. Patients should avoid adding salt to prepared foods and eliminate foods high in sodium (e.g., salt-cured meats, salted snack foods, pickles, soups, delicatessen meats, and processed foods). In patients with hyponatremia (serum Na <130 mEq/L [<130 mmol/L]) or those with persistent volume retention despite high diuretic doses and sodium restriction, daily fluid intake should be limited to 2 L/day from all sources. However, both sodium and fluid restriction must be done with care in patients with HFpEF. Excessive restriction can lead to hypotension, low-output state, and/or renal insufficiency. Daily weights may help to assess volume status. Dietary and lifestyle factors that decrease the risk of development of CAD and HTN should be encouraged.1 Although guidelines recommend reducing sodium intake in patients with HF, proven benefits of such restriction on clinical outcomes are lacking.39 In fact, there is concern among some clinicians about the increased RAAS activity that results from sodium restriction.39
Other important general measures include patient and family counseling on the signs and symptoms of HF, detailed written instructions on the importance of appropriate medication use and compliance, activity level, diet, discharge medications, weight monitoring, continuity of care, and the need for close monitoring and followup to reinforce compliance and minimize the risk of HF exacerbations and subsequent hospitalization. These activities are now referred to as self-care and constitute an important means to improve such important outcomes as hospitalization and quality of life.40
General Approach to Treatment
![]() The ACC/AHA treatment guidelines are organized around the four identified stages of HF, and the treatment recommendations are summarized in Figures 4-7 and 4-8.1 Clinicians are reminded that, in addition to the ACC/AHA, other cardiology professional societies publish guidelines for evaluation and treatment of HF including the Heart Failure Society of America (HFSA) and the European Society of Cardiology (ESC).27,41 Although minor differences exist between the recommendations in these guidelines, they are in general agreement in their overall approach to evaluation and treatment of SHF. In addition to chronic SHF, these guidelines now also provide thorough discussions of acute decompensated HF and management of patients with comorbid diseases often encountered in this population.
The ACC/AHA treatment guidelines are organized around the four identified stages of HF, and the treatment recommendations are summarized in Figures 4-7 and 4-8.1 Clinicians are reminded that, in addition to the ACC/AHA, other cardiology professional societies publish guidelines for evaluation and treatment of HF including the Heart Failure Society of America (HFSA) and the European Society of Cardiology (ESC).27,41 Although minor differences exist between the recommendations in these guidelines, they are in general agreement in their overall approach to evaluation and treatment of SHF. In addition to chronic SHF, these guidelines now also provide thorough discussions of acute decompensated HF and management of patients with comorbid diseases often encountered in this population.
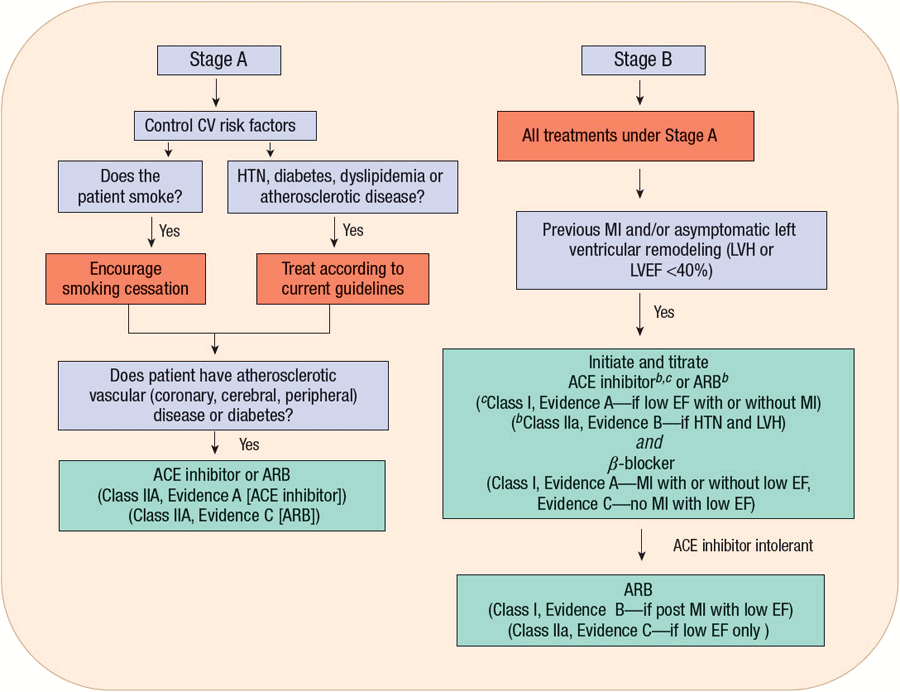
FIGURE 4-7 Treatment algorithm for patients with ACC/AHA Stage A and B heart failure. (Adapted with permission from Hunt SA, Abraham WT, Chin MH, et al. Circulation 2009; 119:e391–e479.)
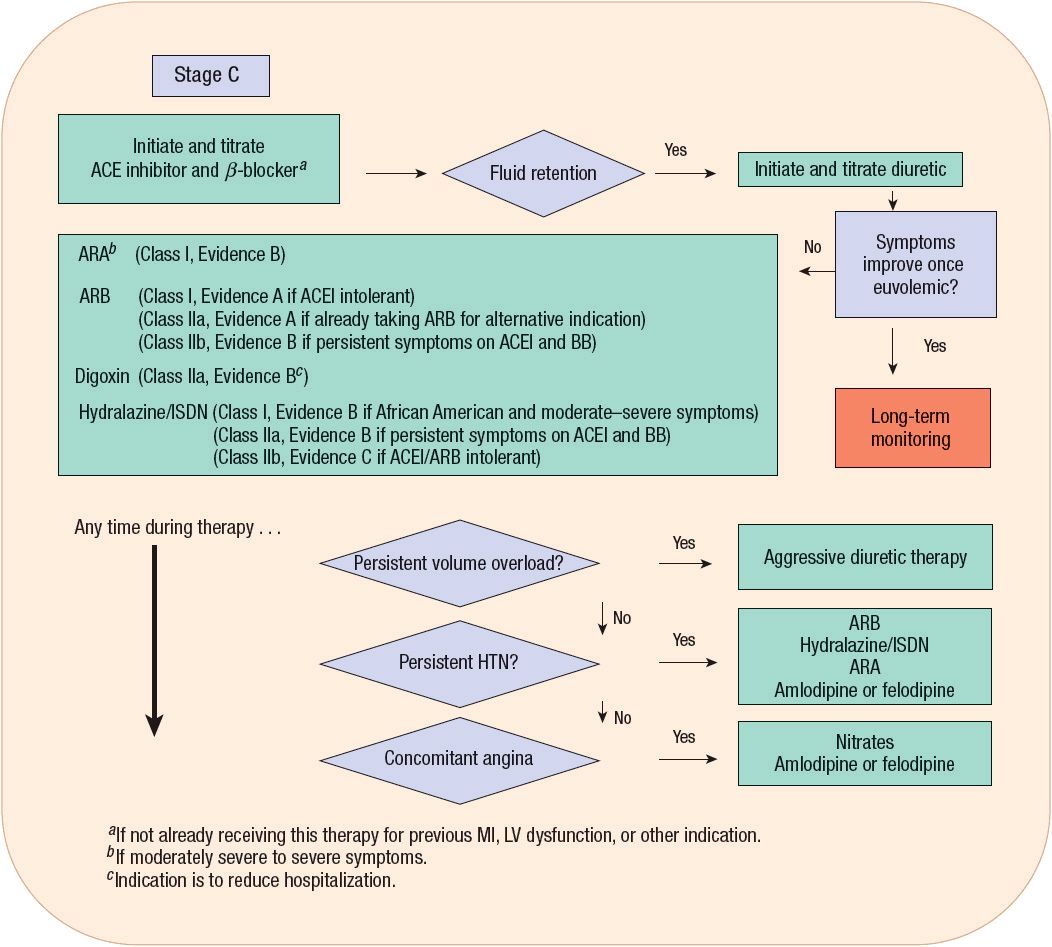
FIGURE 4-8 Treatment algorithm for patients with ACC/AHA Stage C heart failure. (Adapted with permission from Hunt SA, Abraham WT, Chin MH, et al. Circulation 2009; 119:e391–e479.)
Much less objective information on the treatment of HFpEF is available. This relative paucity of evidence is reflected in guidelines for the diagnosis and management of HFpEF published by the ACC/AHA, the ESC, and the HFSA.1,27,41 In general, all three guidelines recommend treating comorbid conditions by controlling HR and BP, alleviating causes of myocardial ischemia, reducing volume, and restoring and maintaining sinus rhythm. Table 4-6 summarizes the therapeutic recommendations from the HFSA.
TABLE 4-6 Evidence-Based Pharmacotherapy for Heart Failure with Preserved Ejection Fraction
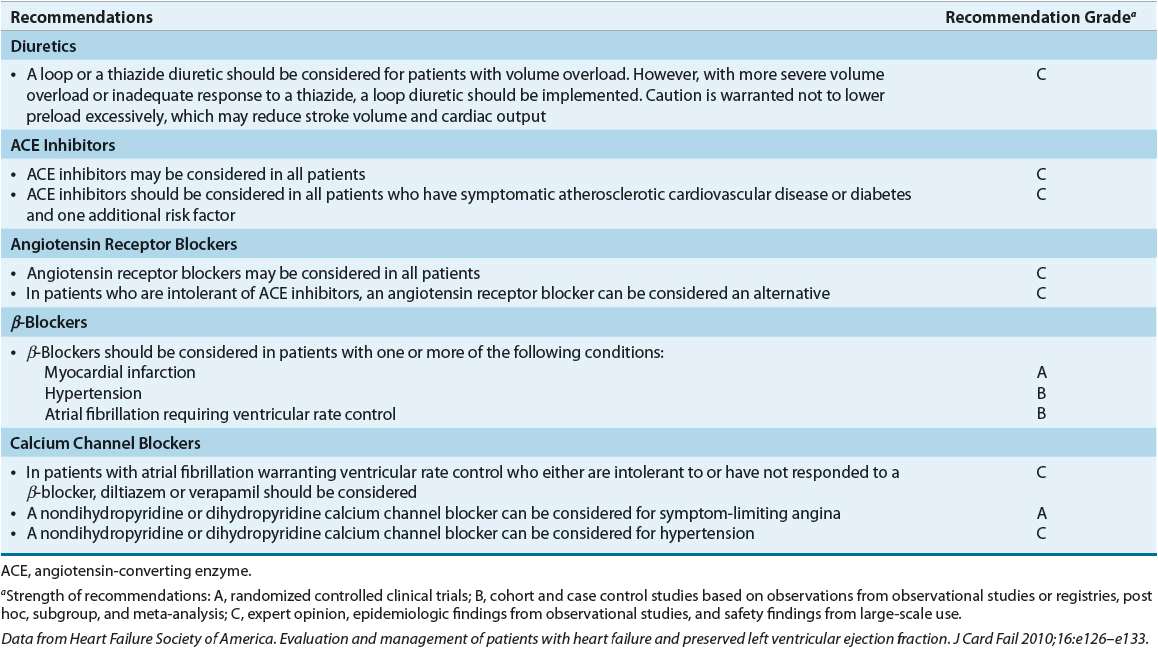
A recent study showing that use of guideline-recommended treatments improves mortality in patients with SHF reinforces the importance for clinicians to be familiar with these recommendations.42 However, clinicians should also remember that these are only guidelines and that evaluation and treatment should be individualized for each patient.
As the management of HF has become increasingly complex, the development of disease management programs that use multidisciplinary teams has been studied extensively. These programs utilize several broad approaches including HF specialty clinics, home-based interventions, structured telephone support, and close patient followup. Most are multidisciplinary and may include physicians, advanced practice nurses, dieticians, and pharmacists. In general, the programs focus on optimization of drug and nondrug therapy, patient and family education and counseling, exercise and dietary advice, intense followup by telephone or home visits, improving adherence to medications and lifestyle recommendations, encouragement of self-care, and early recognition of and management of volume overload.1 Such programs have typically focused on patients with more severe HF who are at high risk for hospital admission. In general, multidisciplinary disease management programs improve quality of life and reduce HF and all-cause hospitalizations and costs, although these benefits are not consistently demonstrated in all studies.43,44
Pharmacists can play an important role in the multidisciplinary team management of HF.34,35,45 Compared with conventional treatment, pharmacist interventions, which included medication evaluation and therapeutic recommendations, patient education, and followup telephone monitoring, reduced hospitalizations for HF, adverse drug events, and medication errors. Pharmacist intervention improved use of disease-modifying medications, although this did not result in better clinical outcomes.46 A recent study found that pharmacist intervention improved medication adherence and reduced emergency department visits and hospitalizations in low-income patients with HF.45 Thus, the benefits of pharmacist involvement in the multidisciplinary care of HF patients are now apparent and should include optimizing doses of HF drug therapy, screening for drugs that exacerbate HF, monitoring for adverse drug effects and drug interactions, educating patients, and patient followup. The role of pharmacists in optimizing pharmacotherapy is underscored by the finding that HF is associated with an increased risk of experiencing adverse drug reactions.47
Treatment of Stage A Heart Failure
Patients in Stage A do not have structural heart disease or HF symptoms but are at high risk for developing HF because of the presence of risk factors (Fig. 4-7). The emphasis here is on risk factor identification and modification to prevent the development of structural heart disease and subsequent HF. Commonly encountered risk factors include HTN, dyslipidemia, diabetes, obesity, metabolic syndrome, smoking, and coronary artery disease. Although each of these disorders individually increases risk, they frequently coexist in many patients and act synergistically to foster the development of HF. Effective control of both systolic and diastolic BPs reduces the risk of developing HF by approximately 50%; thus, current HTN treatment guidelines should be followed.1 Diabetes dramatically increases the risk of developing HF and adversely affects the prognosis of patients with known HF. Appropriate diabetic control is important to minimize the risk of end-organ damage but has not been shown to affect the risk of developing HF. Appropriate management of coronary disease and its associated risk factors is also important.39,48 Although treatment must be individualized, ACE inhibitors or ARBs are recommended for HF prevention in patients with multiple vascular risk factors.1
Treatment of Stage B Heart Failure
Patients in Stage B have structural heart disease, but do not have HF symptoms (Fig. 4-7). This group includes patients with left ventricular hypertrophy, recent or remote MI, valvular disease, or reduced LVEF (less than 40%). These individuals are at risk for developing HF, and treatment is targeted at minimizing additional injury and preventing or slowing the remodeling process. In addition to the treatment measures outlined in Stage A, ACE inhibitors and β-blockers are important components of therapy. Patients with a previous MI should receive both ACE inhibitors and β-blockers, regardless of the LVEF.1 Similarly, patients with a reduced LVEF and no symptoms should also receive both these agents, whether or not they have had an MI.1 ARBs are an effective alternative in patients intolerant to ACE inhibitors.1
Treatment of Stage C Heart Failure
![]()
![]()
![]()
![]()
![]()
![]()
![]() Patients with structural heart disease and previous or current HF symptoms are classified in Stage C (Fig. 4-8). In addition to treatments in Stages A and B, most patients in Stage C should be routinely treated with three medications: a diuretic, an ACE inhibitor, and a β-blocker (see Drug Therapies for Routine Use in Patients with Stage C Systolic Heart Failure below). The benefits of these medications on slowing HF progression, reducing morbidity and mortality, and improving symptoms are clearly established. Aldosterone receptor antagonists, ARBs, digoxin, and hydralazine–isosorbide dinitrate (ISDN) are also useful in selected patients. Nonpharmacologic therapy with devices such as an implantable cardioverter-defibrillator (ICD) or cardiac resynchronization therapy (CRT) with a biventricular pacemaker is also indicated in certain patients in Stage C (see Nonpharmacologic Therapy below). Other general measures noted earlier are also important as is careful followup and patient education to reinforce dietary and medication compliance to prevent clinical deterioration and reduce hospitalization.1,40
Patients with structural heart disease and previous or current HF symptoms are classified in Stage C (Fig. 4-8). In addition to treatments in Stages A and B, most patients in Stage C should be routinely treated with three medications: a diuretic, an ACE inhibitor, and a β-blocker (see Drug Therapies for Routine Use in Patients with Stage C Systolic Heart Failure below). The benefits of these medications on slowing HF progression, reducing morbidity and mortality, and improving symptoms are clearly established. Aldosterone receptor antagonists, ARBs, digoxin, and hydralazine–isosorbide dinitrate (ISDN) are also useful in selected patients. Nonpharmacologic therapy with devices such as an implantable cardioverter-defibrillator (ICD) or cardiac resynchronization therapy (CRT) with a biventricular pacemaker is also indicated in certain patients in Stage C (see Nonpharmacologic Therapy below). Other general measures noted earlier are also important as is careful followup and patient education to reinforce dietary and medication compliance to prevent clinical deterioration and reduce hospitalization.1,40
Dozens of trials evaluated pharmacotherapy in patients with SHF, but few focused on patients with HFpEF. In fact, most published HF studies specifically excluded patients with preserved EF. The results of large clinical trials for treatment of HFpEF as well as important ongoing studies are summarized in Table 4-7. The hope is that these studies will provide the future basis for evidence-based treatment for HFpEF.
TABLE 4-7 Completed and Ongoing Large Clinical Trials for HFpEF
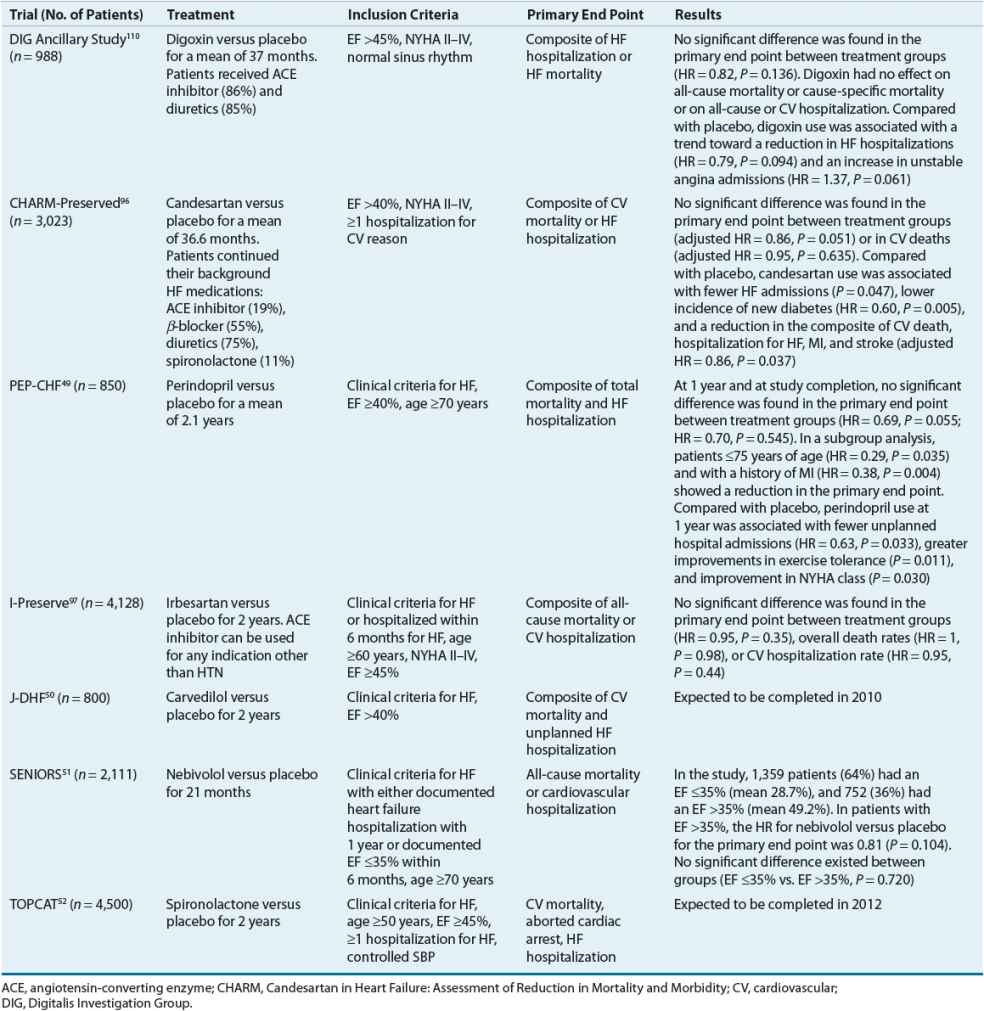
Treatment of Stage D Heart Failure
Stage D HF includes patients with refractory symptoms at rest despite maximal medical therapy and those patients who undergo recurrent hospitalizations or cannot be discharged from the hospital without special interventions. These individuals have the most advanced form of HF and should be referred to HF management programs so that specialized therapies including mechanical circulatory support, continuous IV positive inotropic therapy, and cardiac transplantation can be considered in addition to standard treatments outlined in Stages A to C.1 Discussions with the patient and family members regarding prognosis, patient priorities for minimizing symptoms versus prolonging survival, options for additional treatments, and end-of-life care should be started.1,53 A scientific statement from the AHA on decision making in advanced HF is an excellent resource on these issues.54
Management of volume status can be challenging in these patients. Restriction of sodium and fluid intake may be beneficial. High doses of diuretics, combination therapy with a loop and thiazide diuretic, or mechanical methods of fluid removal such as ultrafiltration may be required. Patients in Stage D may be less tolerant to ACE inhibitors (hypotension, worsening renal insufficiency) and β-blockers (worsening HF) as high levels of neurohormonal activation maintain circulatory homeostasis. Initiation of therapy with low doses, slow upward dose titration, and close monitoring for signs and symptoms of intolerance are essential in this group of patients. The approach to treatment of patients with Stage D HF is discussed in more detail in Chapter 5.
Nonpharmacologic Therapy
Sudden cardiac death, primarily due to ventricular tachycardia and fibrillation, is responsible for 40% to 50% of the mortality in patients with HF. In general, patients in the earlier stages with milder symptoms are more likely to die from sudden death, whereas death from pump failure is more frequent in those with advanced HF. Many of these patients have complex and frequent ventricular ectopy, although it remains unknown whether these ectopic beats contribute to the risk of malignant arrhythmias or merely serve as markers for individuals at higher risk for sudden death. Drugs that attenuate disease progression such as β-blockers and aldosterone antagonists reduce the risk of sudden death. However, although class I antiarrhythmic agents can suppress ventricular ectopy, empiric treatment with them adversely affects survival.55
Implantation of an ICD is an effective primary prevention for reducing the risk of mortality from sudden death.1 In patients with NYHA class II or III symptoms and LVEF ≤35%, the ICD was superior to amiodarone or placebo for reducing mortality.56 Importantly, this study also found that amiodarone was no more effective than placebo. Thus, this drug, because of its multiple adverse effects, drug interactions, and lack of effect on mortality, should not be used for primary prevention of sudden death. However, because of the neutral effects of amiodarone on survival, it is often used in HF patients with symptomatic atrial fibrillation to maintain sinus rhythm and/or to prevent ICD discharges. The ACC/AHA guidelines recommend the ICD for both primary and secondary prevention to improve survival in patients with current or previous HF symptoms and reduced LVEF.1 A thorough review of ICD therapy can be found in Chapter 8.
The use of CRT improves a number of important end points in selected patients with chronic SHF.57,58 Delayed electrical activation of the left ventricle, characterized on the ECG by a QRS duration that exceeds 120 milliseconds, occurs in approximately one third of patients with moderate to severe SHF. Since the left and right ventricles normally activate simultaneously, this delay results in asynchronous contraction of the ventricles, which contributes to the hemodynamic abnormalities of HF. Implantation of a specialized biventricular pacemaker to restore synchronous activation of the ventricles improves ventricular contraction and hemodynamics. Use of CRT is associated with improvements in exercise capacity, NYHA classification, quality of life, hemodynamic function, hospitalizations, and mortality.57,58 The ACC/AHA guidelines recommend CRT in NYHA class III to IV patients receiving optimal medical therapy and with a QRS duration ≥120 milliseconds and LVEF ≤35%.1 However, trials published since the last guideline update show impressive benefits in patients with less severe symptoms (NYHA class I to III) and that this earlier use of CRT is associated with reverse remodeling.46,57,59 It is likely that these findings will be incorporated into the next guideline revision. Combined CRT and ICD devices are available and can be used if the patient meets the indications for both devices.
Pharmacologic Therapy
![]() With a few notable exceptions, many of the drugs used to treat SHF are the same as those for treatment of HFpEF. However, the rationale for their use, the pathophysiologic process that is being altered by the drug, and the dosing regimen may be entirely different depending on whether the patient has SHF or HFpEF. For example, β-blockers are recommended for the treatment of both SHF and HFpEF. In HFpEF, however, β-blockers are used to decrease HR, increase diastolic duration, and modify the hemodynamic response to exercise. In SHF, β-blockers are used in the long term to increase the inotropic state and modify LV remodeling. Diuretics also are used in the treatment of both SHF and HFpEF. However, the doses of diuretics used to treat HFpEF are, in general, much smaller than those used to treat SHF. Antagonists of the RAAS are useful in lowering BP and reducing LVH. Some drugs, however, are used to treat either SHF or HFpEF, but not both. Calcium channel blockers such as diltiazem, nifedipine, and verapamil have little utility in the treatment of SHF. In contrast, each of these drugs has been proposed as being useful in the treatment of HFpEF.
With a few notable exceptions, many of the drugs used to treat SHF are the same as those for treatment of HFpEF. However, the rationale for their use, the pathophysiologic process that is being altered by the drug, and the dosing regimen may be entirely different depending on whether the patient has SHF or HFpEF. For example, β-blockers are recommended for the treatment of both SHF and HFpEF. In HFpEF, however, β-blockers are used to decrease HR, increase diastolic duration, and modify the hemodynamic response to exercise. In SHF, β-blockers are used in the long term to increase the inotropic state and modify LV remodeling. Diuretics also are used in the treatment of both SHF and HFpEF. However, the doses of diuretics used to treat HFpEF are, in general, much smaller than those used to treat SHF. Antagonists of the RAAS are useful in lowering BP and reducing LVH. Some drugs, however, are used to treat either SHF or HFpEF, but not both. Calcium channel blockers such as diltiazem, nifedipine, and verapamil have little utility in the treatment of SHF. In contrast, each of these drugs has been proposed as being useful in the treatment of HFpEF.
Drug Therapies for Routine Use in Patients with Stage C Systolic Heart Failure
![]()
![]()
![]()
![]() A treatment algorithm for management of patients with Stage C SHF is shown in Figure 4-8. In general, these patients should receive combined therapy with an ACE inhibitor or ARB (if ACE inhibitor intolerant) and a β-blocker, plus a diuretic if there is evidence of fluid retention. An aldosterone receptor antagonist should also be considered in selected patients.1 Initiation of digoxin therapy can be considered at any time for symptom reduction, to decrease hospitalizations, or slow ventricular response in patients with concomitant atrial fibrillation.1 Drug dosing and monitoring are summarized in Tables 4-8 and 4-9.
A treatment algorithm for management of patients with Stage C SHF is shown in Figure 4-8. In general, these patients should receive combined therapy with an ACE inhibitor or ARB (if ACE inhibitor intolerant) and a β-blocker, plus a diuretic if there is evidence of fluid retention. An aldosterone receptor antagonist should also be considered in selected patients.1 Initiation of digoxin therapy can be considered at any time for symptom reduction, to decrease hospitalizations, or slow ventricular response in patients with concomitant atrial fibrillation.1 Drug dosing and monitoring are summarized in Tables 4-8 and 4-9.
TABLE 4-8 Drug Dosing Table