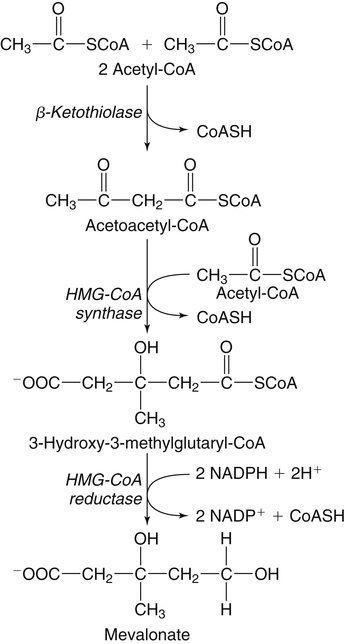
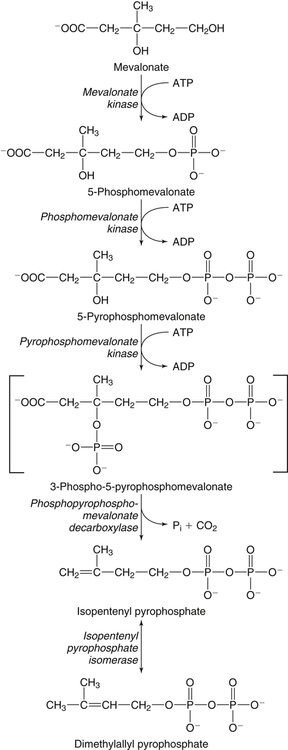
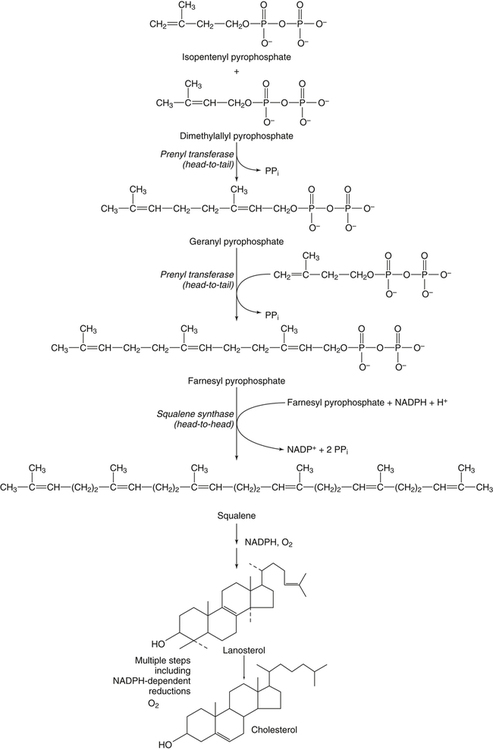
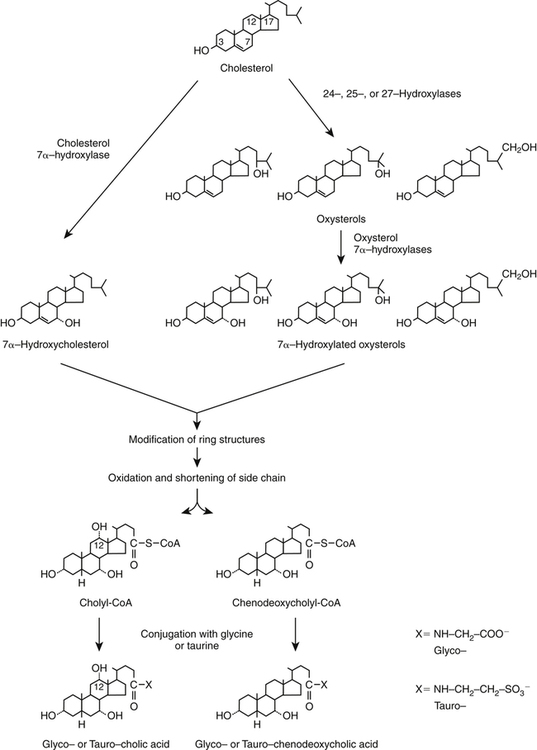
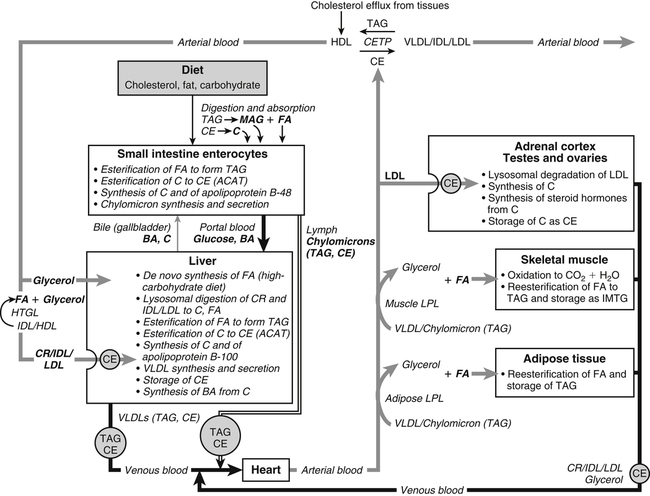
Hei Sook Sul, PhD and Judith Storch, PhD Cholesterol is a sterol molecule with critical biological importance. It is an essential structural component of cell membranes, affecting their stability and permeability. Cholesterol also is essential as the precursor of the sex and adrenal steroid hormones and of the bile acids that play important roles in lipid digestion and absorption. Oxidized metabolites of cholesterol, called oxysterols, are also important regulators of lipid metabolic pathways. Moreover, isoprenoids, intermediates in the cholesterol biosynthetic pathway, are used for production of small amounts of molecules that have important biological functions. These include dolichol, which is required for glycoprotein synthesis, and ubiquinone (coenzyme Q), which is involved in electron transport in mitochondria. Isoprenoids also are used for posttranslational farnesylation or geranylgeranylation (called prenylation) of a variety of proteins including the membrane-associated heterotrimeric G proteins and the small GTP-binding proteins. Cholesterol itself is also used for modification of specific proteins. The covalently attached isoprenoid or cholesterol moieties serve in most cases as anchors for targeting proteins to certain membrane compartments or submembrane domains. An overview of major aspects of cholesterol metabolism and transport by lipoproteins is shown in Figure 17-1. Cholesterol is transported in the plasma lipoprotein fractions. Transport of dietary cholesterol to the tissues is mediated initially by chylomicrons and subsequently by very-low-density lipoproteins (VLDLs) after the liver reincorporates cholesterol from chylomicron remnants into these new lipoprotein particles. Tissues remove triacylglycerol from lipoproteins, as discussed in Chapter 16, converting them into lipoprotein remnants that are removed by the liver. Much of the VLDL remnants (also called intermediate-density lipoproteins, or IDLs) are further metabolized to cholesterol-rich low-density lipoproteins (LDLs), which are taken up by most tissues as a source of cholesterol. Cholesterol excretion largely occurs via cholesterol efflux to form high-density lipoproteins (HDLs) which deliver cholesterol to tissues through exchange of cholesteryl esters for triacylglycerol molecules from other lipoprotein fractions, ultimately transferring the cholesterol to the LDL pool. LDL is mainly removed by the liver, and the liver secretes both cholesterol and bile acids synthesized from cholesterol into the lumen of the small intestine via the bile. To the extent that the cholesterol and bile acids are not completely reabsorbed, there is some net excretion of cholesterol in the feces. Each of these steps in cholesterol metabolism is discussed in this chapter. The first steps in cholesterol biosynthesis result in the synthesis of mevalonate, a 6-carbon compound, from acetyl-CoA, as shown in Figure 17-2. Synthesis of the key intermediate 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) follows the same pathway described in Chapter 16 for the synthesis of ketone bodies, except that it occurs in the cytosol instead of the mitochondria, catalyzed by enzymes localized to the ER membrane. Two molecules of acetyl-CoA condense to form acetoacetyl-CoA (acetyl-CoA:acetoacetyl-CoA acetyltransferase). Another molecule of acetyl-CoA then condenses with a molecule of acetoacetyl-CoA to form HMG-CoA, a reaction catalyzed by HMG-CoA synthase. HMG-CoA is converted to mevalonate (see Figure 17-1) by HMG-CoA reductase, which is anchored in the ER membrane with its active site located in a long carboxyl terminal domain in the cytosol: Conversion of mevalonate to “active” isoprenoid units (5-carbon units) occurs as summarized in Figure 17-3. Mevalonate is converted to 3-phospho-5-pyrophosphomevalonate by three sequential phosphorylations. Squalene, and thus the sterol molecule, is built from multiple isopentenyl groups, as shown in Figure 17-4. The reaction sequence involves condensation of two 5-carbon molecules to form one of 10 carbons. A third 5-carbon molecule is added to form a 15-carbon intermediate. Two 15-carbon intermediates are linked to form the 30-carbon squalene: Formation of the 10-carbon geranyl pyrophosphate involves two enzymatic reactions. First, isopentenyl pyrophosphate isomerase catalyzes the isomerization of isopentenyl pyrophosphate to dimethylallyl pyrophosphate (as shown in Figure 17-3). Then one molecule of each of these two activated isoprenes condenses. As shown in Figure 17-4, C1 of one isoprene bonds with C5 of the other (head-to-tail) to form geranyl pyrophosphate. In Chapter 16, we noted that activation of fatty acids to their acyl-CoA derivatives involves hydrolysis of ATP to AMP and PPi. Ubiquitous pyrophosphatases in cells then rapidly degrade PPi to two inorganic phosphates. This same biochemical mechanism is used to drive to completion several of the intermediate reactions in isoprenoid and cholesterol biosynthesis. Cholesterol itself regulates transcription of HMG-CoA reductase, as well as other enzymes involved in cholesterol homeostasis, by a mechanism that senses cellular cholesterol levels (Brown and Goldstein, 2009). Transcriptional control by cholesterol requires the presence of a sterol regulatory element (SRE) in the promoter regions of the genes where the transcription factor termed SRE-binding protein (SREBP) binds. There are three SREBPs: SREBP1a and SREBP1c, which are both encoded by the SREBP1 gene by alternative exon usage, and SREBP2, which is encoded by the SREBP2 gene. SREBPs induce transcription of genes coding for the enzymes involved in not only cholesterol metabolism but also fatty acid and triacylglycerol synthesis. SREBP2 is the main form that responds to cholesterol levels and activates transcription of genes encoding proteins involved in cholesterol metabolism including all the enzymes in cholesterol biosynthesis and the LDLR. SREBP1a activates genes involved in both cholesterol and fatty acid synthesis and thus may be important during cell division in meeting the need for increased membrane lipids. In contrast, SREBP1c expression is increased by insulin and/or feeding. SREBP1c mediates the induction of enzymes in fatty acid and triacylglycerol synthesis, as described in Chapter 16. All SREBPs contain two transmembrane helices that anchor these proteins in the ER. The N-terminal domain faces the cytosol and contains a basic helix-loop-helix leucine zipper transcription factor motif. When cholesterol level in the ER membrane is high, cholesterol directly binds SCAP, triggering SCAP to bind to an ER retention protein called Insig (insulin-induced gene). In addition, oxysterols, which are oxygenated metabolites of cholesterol present at low concentrations that are produced as part of the cholesterol excretion pathway as described below, directly bind to Insig, triggering Insig to bind to SCAP. In both cases, the end result is a conformational change in SCAP that prevents its binding to COPII coat proteins, consequently preventing vesicular transport of the SCAP/SREBP complex to the Golgi apparatus and thereby decreasing the release of SREBP. Thus SREBP cannot be transported to the nucleus, and the transcription of genes mediating cholesterol biosynthesis and uptake is not enhanced (Figure 17-5). In addition to regulation of enzyme protein levels, the activity of HMG-CoA reductase is regulated by a phosphorylation–dephosphorylation mechanism. HMG-CoA reductase is less active in the phosphorylated state. The enzyme is phosphorylated by AMP-activated protein kinase (AMPK), a kinase that is activated by an increase in cellular AMP as well as by phosphorylation by upstream kinases, such as LKB1 (Steinberg and Kemp, 2009). This phosphorylation–dephosphorylation mechanism explains why HMG-CoA reductase activity can respond rapidly to nutritional or hormonal perturbations, even when there has been little change in the concentration of HMG-CoA reductase protein. Because cholesterol is a hydrophobic molecule, almost all of it is found in membranes or lipid droplets, and little is found in the soluble fraction. Most of the cellular free cholesterol (60% to 80%) is found in the plasma membrane. Excess cholesterol may be esterified to fatty acids and stored in lipid droplets. Cholesterol synthesized within the cell originates in the ER where it is synthesized, whereas cholesterol taken up from the plasma is brought into the endosomal–lysosomal system via LDL receptor–mediated endocytosis. Within the cell, the subcellular distribution of cholesterol is regulated by several distinct processes. There are two general mechanisms for intracellular cholesterol trafficking (Soccio and Breslow, 2004). One mechanism is through vesicular transport, in which cholesterol incorporated into membranes moves as part of membrane vesicles. The second way in which cholesterol can move from one subcellular location to another is by the monomolecular diffusion of protein-bound, or perhaps unbound, cholesterol, within the aqueous cytosol. Distinct trafficking pathways exist for cholesterol synthesized de novo and for cholesterol obtained via receptor-mediated endocytosis of LDLs. Specific proteins in the LE/LY compartment are thought to be involved in the egress of cholesterol from the vesicular compartment. Evidence for a role of the Niemann-Pick type C (NPC) disease proteins (NPC1 and NPC2) in cholesterol trafficking or transport comes from abnormalities observed in patients with NPC disease. NPC disease is caused by defects in either NPC1 or NPC2, and the indistinguishable clinical phenotypes produced by defects in either protein suggest that the two proteins work together. In patients with NPC disease, unesterified cholesterol, as well as glycolipids, are trapped in the LE/LY compartment. Progression of neurological deterioration and premature death in these patients are thought to be due to both derangement in LE/LY function, which is secondary to sterol and lipid accumulation, and the absence of normal postendolysosomal metabolism of cholesterol. The transporter NPC1 has multiple membrane-spanning domains and contains a sterol-sensing domain similar to that found in several other proteins involved in cholesterol homeostasis, including SCAP and HMG-CoA reductase. NPC2 is a soluble intralysosomal protein that binds cholesterol. It has been proposed that NPC2 binds cholesterol directly following its generation from cholesterol esters by acid lipase; or it may extract membrane-bound cholesterol from internal LE/LY membranes by interacting with the LE/LY−specific phospholipid, lysobisphosphatidic acid (Storch and Xu, 2009). The cholesterol bound to NPC2 in the LE/LY lumen is proposed to then be delivered to NPC1, residing in the limiting membrane of the LE/LY, so that it can then be exported from the LE/LY compartment to other organelles (Wang et al., 2010; Peake and Vance, 2010). Cholesterol from the LE/LY compartment is trafficked primarily to the plasma membrane, with some of the cholesterol going through the Golgi apparatus before reaching the plasma membrane. A small portion of exogenously derived cholesterol moves from the LE/LY compartment directly to the ER. At the inner mitochondrial membrane of steroidogenic cells, cholesterol is first converted into pregnenolone, the precursor of all steroid hormones. These reactions occur on the inner leaflet of the inner mitochondrial membrane. Mitochondrial cholesterol may be trafficked from the plasma membrane, from the endosomal–lysosomal compartment, or perhaps from lipid droplets. The movement of cholesterol into the mitochondria is accomplished by steroidogenic acute regulatory protein (StAR), which interacts with cholesterol and with specific outer mitochondrial membrane proteins in assembling a transport complex that moves the lipid to the inner mitochondrial membrane. The principal protein that interacts with StAR is translocator protein, TSPO (previously called peripheral-type benzodiazepine receptor). Cholesterol is passed from StAR to TSPO, which, along with other interacting proteins, moves cholesterol from the outer to the inner mitochondrial membrane (Rone et al., 2009; Mesmin and Maxfield, 2009). As mentioned in Chapter 7, conversion of cholesterol to bile acids occurs exclusively in the liver. Bile acids and free cholesterol are incorporated into bile, which is destined for secretion into the proximal small intestine where the bile acids facilitate digestion and absorption of lipids. The ultimate excretion of bile acids and free cholesterol in bile is the major route for removing excess cholesterol from the body. Bile acids also have endocrine functions; they are ligands for several nuclear receptors that modulate a variety of cellular processes (Lefebvre et al., 2009). Synthesis of bile acids involves a cascade of reactions catalyzed by enzymes located in the ER, mitochondria, cytosol, and peroxisomes. The steroid nucleus is modified in the ER and mitochondria, and the cholesterol side chain is removed in peroxisomes. The major steps of the two pathways for bile acid synthesis are shown in Figure 17-6 (Russell, 2009). In the major or “classic” pathway, the steroid nucleus modification includes 7α-hydroxylation catalyzed by cholesterol 7α-hydroxylase (CYP7A1), epimerization of the 3β-hydroxyl group (to 3α-), and saturation of the steroid nucleus. There is also a 12α-hydroxylation in the case of cholic acid synthesis, but not in chenodeoxycholic acid synthesis. These reactions precede the removal of the terminal three carbons of the cholesterol side chain by oxidative cleavage. In an alternative pathway, hydroxylation of the side chain at either the C24, C25, or C27 position to form oxysterols precedes the modification of the steroid nucleus by 7α-hydroxylation. Cholic acid and chenodeoxycholic acid are the two primary bile acids in human bile. They are conjugated with either glycine or taurine to become water soluble and are then secreted from the liver into the bile. Further modification of the primary bile acids by gut bacteria form secondary bile acids, which may be reabsorbed via the enterohepatic circulation and are thus found in the liver and bile along with the primary bile acids. The rate of bile acid synthesis parallels the activity of CYP7A1, the main regulated step in the bile acid synthetic pathway. This step is under feedback regulation. Bile acids excreted from the liver are reabsorbed in the distal small intestine and transported back to the liver by the enterohepatic recirculation as mentioned below and in Chapter 7 (see Figure 7-9). Therefore bile acid reabsorption controls the overall rate of bile acid synthesis in the liver. Identification of farnesoid X receptor (FXR), which belongs to the nuclear receptor family, as a bile acid receptor provides a mechanism for this feedback regulation (Hageman et al., 2010). Thus gene expression for several proteins in bile acid metabolism, such as CYP7A and ileal BABP (an intestinal bile acid–binding protein that may shuttle bile acids from the apical to basolateral side of enterocytes upon reabsorption), may be controlled by FXR, either directly or indirectly. Bile acids may also regulate lipid, glucose, and energy metabolism by binding not only to FXR but also to the other nuclear hormone receptors, such as pregnane X receptor (PXR), constitutive androstane receptor (CAR), and a G protein–coupled bile acid receptor (TGR5). Oxysterols, potent regulators of cholesterol synthesis and lipid metabolism, can be formed from cholesterol by both enzymatic and nonenzymatic reactions (Bjorkhem, 2009). The most abundant oxysterol in human plasma is 27-hydroxycholesterol. Oxysterols are ligands for another member of the nuclear hormone receptor family, liver X receptor (LXR) (Calkin and Tontonoz, 2010). The LXR response element is present in the promoter region of the CYP7A1 gene, and LXR binding activates CYP7A1 gene transcription in mice. Therefore regulation of CYP7A1 transcription and bile acid synthesis in some mammals may depend on the balance between the positive LXR control and negative FXR control. The human CYP7A1 promoter, however, does not contain an LXR-binding site and may not be a direct target of LXR. Nevertheless, LXR activates a variety of other genes involved in cholesterol and triacylglycerol metabolism. LXR activates transcription of genes encoding ATP-binding cassette transporters and other proteins involved in reverse cholesterol transport, mentioned later in this chapter. In addition, activation of the SREBP1c gene by LXR stimulates transcription of enzymes in fatty acid and triacylglycerol synthesis. Newly synthesized bile acids, along with those transported into the liver from the portal blood that drains the gastrointestinal tract, are secreted into bile for storage in the gallbladder before release into the small intestine after a meal (see Chapters 7 and 10). Cholesterol is also a component of bile, and secretion of cholesterol into bile is facilitated by the heterodimer of ABCG5/ABCG8, present at the canalicular membrane of hepatocytes. ABCG5/ABCG8 is also present on the brush border membrane of enterocytes where it secretes sterols in enterocytes back into the intestinal lumen. The rare mutation of either ABCG5 or ABCG8 causes β-sitosterolemia. Normally, plant sterol levels in plasma and tissues are very low due to the secretion of plant sterols back into the gut lumen. Patients with β-sitosterolemia absorb plant sterols efficiently, resulting in abnormally high plant sterol levels (including β-sitosterol) in plasma and tissues and deposition of sterols in the skin and arteries, which increases the risk of premature atherosclerosis (Berge et al., 2000). Solubilization of biliary and dietary sterols in the intestinal lumen is aided by bile acids and phospholipids to form mixed micelles. Sterols are then absorbed via Niemann-Pick C1–like 1 protein (NPC1L1) at the apical membrane of enterocytes (Davis and Altmann, 2009; Betters and Yu, 2010). Ezetimibe, a plasma cholesterol reducing drug, inhibits intestinal absorption of cholesterol by binding to NPC1L1. Ezetimibe is used in combination with statins for lowering plasma cholesterol levels.
Cholesterol and Lipoproteins: Synthesis, Transport, and Metabolism
Overview of Cholesterol Metabolism
Synthesis of Cholesterol and Isoprenoids
Cholesterol Synthesis: Conversion of Acetyl-CoA to Mevalonate in the Cytosol
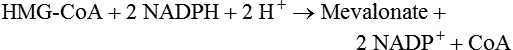
Cholesterol Synthesis: Synthesis of Squalene and Isoprenoids from Mevalonate
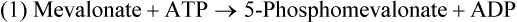


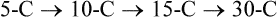
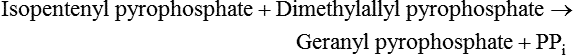
Regulation of Cholesterol Synthesis
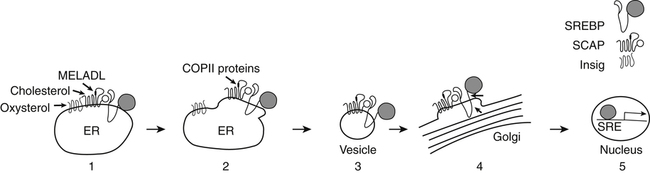
Role of Sterol Regulatory Element–Binding Protein in Transcriptional Responses to Changes in Cholesterol Levels
High Cellular Cholesterol
Regulation of HMG-CoA Reductase by Phosphorylation
Intracellular Trafficking of Cholesterol Synthesized in Cell Versus Cholesterol taken up from Plasma in LDL
Synthesis of Steroid Hormones from Cholesterol
Synthesis of Bile Acids and Oxysterols from Cholesterol
Cholesterol Transport During Enterohepatic Recirculation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


