- Chloramphenicol has a broad spectrum of activity but, due to toxicity issues, its use is reserved for severe infections or for topical applications.
- Chloramphenicol inhibits protein synthesis through binding to the large ribosome subunit (50S) at the peptidyl transferase centre A site, thus preventing binding of the next charged tRNA.
- Resistance arises due to chloramphenicol acetyltransferases (CATs), which acetylate chloramphenicol so that it no longer binds to this site.
4.4.1 Discovery
Chloramphenicol was first obtained from a Streptomyces species isolated from the filtrate of a soil sample from a field near Caracas (Ehrlich et al., 1947). For obvious reasons, the bacterium was named Streptomyces venezuelae and, once it had been established that this crystalline antibiotic contained covalently-linked chlorine, it was originally named chloromycetin. Chloramphenicol (Chloromycetin, Cm) was soon found to be bacteriostatic and to have a broad spectrum of activity.
4.4.2 Synthesis
Unlike some of the other antibiotics we will study in this text, chloramphenicol has a relatively simple structure, so that, although it was originally obtained from fermentation, once its structure had been deduced and the active diastereoisomer identified, chloramphenicol could be produced by total synthesis. As chloramphenicol contains two stereogenic (chiral) centres, it has four possible stereoisomers,17 with only the R,R-diastereoisomer (shown in Figure 4.4.1) of the four having antibacterial activity. Any synthetic method thus needs to produce this isomer, preferably with as few steps as possible involving the separation of isomers, as this is both costly and wasteful (if a synthetic step produces a pair of enantiomers which need to be separated then 50% of the product will be discarded after the enantiomeric resolution18). The first synthesis of chloramphenicol was reported in 1949 and involved eight steps, including an enantiomeric resolution in the second-last step (a particularly wasteful process as 50% of the racemate is discarded after having been carried through the previous six steps) (Controulis et al., 1949). In addition, the original work describes an explosion which occurred in one of the earlier steps, so it is probably best not to consider this route to chloramphenicol in detail and to look instead at a recent example of the many stereoselective syntheses (Scheme 4.4.1). Among the attractive features of this particular route is that it is short (only three steps and an overall yield of 23% from 4-nitrobenzaldehyde) and gives the desired, (−)-chloramphenicol, enantiomer in high purity, as the epoxide intermediate is formed in 95% enantiomeric excess (i.e. a ratio of 97.5 : 2.5 in favour of the enantiomer shown) by the Sharpless asymmetric epoxidation (K. B. Sharpless, Nobel Prize in Chemistry, 2001). The subsequent steps are all stereospecific, with the lone pair of the imidate nitrogen attacking the epoxide from the side opposite to the oxygen of the ring (in order to minimise the repulsion between this lone pair and those on the oxygen), resulting in an inversion of the stereochemistry at this centre, from (S) to (R) (Bhaskar et al., 2004).
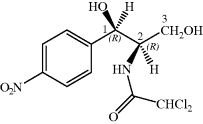
Scheme 4.4.1 Stereoselective synthesis of chloramphenicol ((−)-DIPT = (−)-diisopropyl tartrate, iPrO2C·CHOH·CHOH·CO2iPr)
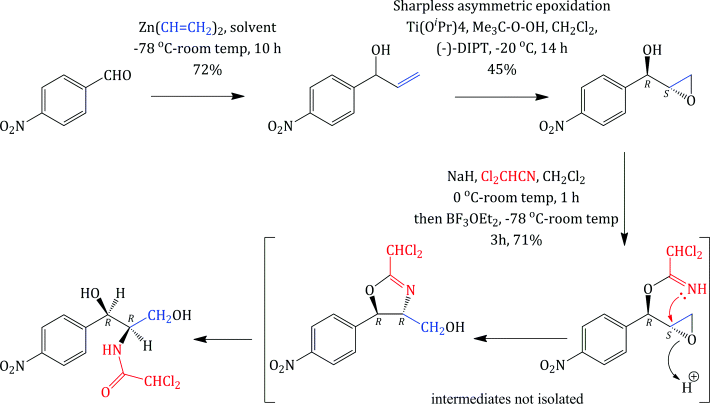
Table 4.4.1 Therapeutic indications for chloramphenicol.
| Indication | Notes |
| Typhoid fever | Where use of quinolones is unsuitable |
| Meningitis | When patient is allergic to penicillins or cephalosporins |
| Epiglottitis | Given intravenously |
| Eye and ear infections | Topical application |
4.4.3 Bioavailability
When you look again at the structure of chloramphenicol, you can see that, compared to some of the other antibiotics we have encountered in this section, it is a relatively small molecule. Having two hydroxyl functional groups (which are often responsible for aiding aqueous solubility), you might think that it would be relatively water-soluble. Aromatic nitro compounds, however, are well known for their insolubility, particularly in aqueous solutions, and chloramphenicol is no exception – it has low aqueous solubility (~2.5 g/L at 25 °C and neutral pH), with its lowest solubility occurring at pH 5.5–7.0 (i.e. in the middle of the physiologically relevant pH range). Amide NH groups are usually considered to be neutral, but that of chloramphenicol is significantly more acidic than most, due to the electronegative influence of the two chlorine atoms causing greater than usual electron flow from the amide N to the carbonyl group. However, although it is more acidic, the pKa of 11.0319 (Qiang and Adams, 2004) does not allow for its ready ionisation. The low solubility of chloramphenicol was sufficiently limiting to its clinical use that two prodrug forms were developed; chloramphenicol succinate (as the ionised sodium salt), with high aqueous solubility (suitable for IV administration, for example in an emergency situation), and chloramphenicol palmitate, which is essentially water-insoluble and designed for oral administration (Figure 4.4.2). A comparison of the relative bioavailability of these prodrugs showed that the orally administered palmitate has a greater and more predictable bioavailability in infants and children (Pickering et al., 1980; Kauffmann et al., 1981).
Chloramphenicol itself is stable, with a shelf life of several years for the crystalline form, and it retains 100% of its activity for 24 hours in solution over a pH range of 1–9.5 (Bartz, 1948); however, over a longer time period, it degrades in solution due to exposure to light (Zhou et al., 2010), heat (Franje et al., 2010), and different pHs (Boer and Pijnenburg, 1983; Zhou et al., 2010). The succinate prodrug also suffers from instability in solution over 1 week, even when stored at −20 °C; the temperature must be decreased to −50 °C to achieve good stability over 1 month (Ambrose, 1984).
Chloramphenicol succinate is readily ionised, with a pKa of 5.5 (Reynolds, 1993), and is usually administered as its sodium salt. It has unpredictable bioavailability, as the prodrug is not reliably hydrolysed to form chloramphenicol, and a variable amount (dependent upon factors such as patient group and liver and kidney function) of the prodrug is excreted unchanged in the urine (Nahata and Powell, 1981, 1983; Ambrose, 1984 and references therein). As a result, the plasma levels of chloramphenicol must be monitored, especially in neonates, infants, children, elderly patients, and those with impaired liver and kidney function (Ambrose, 1984; Wenk et al., 1984; Weber et al., 1999). The most important bioavailability parameters for parenterally administered chloramphenicol succinate are the rate of esterase hydrolysis to active drug, balanced against the renal excretion of the prodrug (Ambrose, 1984).
Chloramphenicol palmitate, the water-insoluble prodrug, is insoluble in saliva and so does not bind to the taste receptors on the tongue, and was developed for oral use to overcome the very unpleasant extreme bitter taste of chloramphenicol. It is hydrolysed by esterase activity in the small intestine, releasing chloramphenicol to be absorbed from the GI tract. When administered orally as a suspension, the chloramphenicol palmitate prodrug has a bioavailability of around 80% and reaches peak plasma concentrations after about 2–3 hours (Ambrose, 1984). An oral suspension of chloramphenicol palmitate is used for paediatric applications in some countries, but not currently in the UK.
Whether delivered IV or orally, once released, about 10% of chloramphenicol is excreted unchanged in the urine, while 90% is metabolised to inactive products before renal excretion (Ambrose, 1984). The major metabolic pathway is that of glucuronidation, forming two products: the 3-O-glucuronide (major product, formed by glucuronidation on the less hindered primary alcohol) and the 1-O-glucuronide (minor product, from glucuronidation on the secondary alcohol) (Chen et al., 2007, 2010). Other minor metabolites may arise due to hydrolysis of the amide and reduction of the aromatic nitro group; the aromatic nitroso and hydroxylamine reduction products may cause toxicity, and this is discussed further in Subsection 4.4.7. The half-life of chloramphenicol depends upon the mode of administration, t½ being 0.6–2.7 hours for chloramphenicol succinate and 2.3–4.0 hours for chloramphenicol. It is widely distributed throughout many tissues and compartments, with a Vd (volume of distribution) of 0.57 L/kg for adults with normal hepatic and renal function (Ghafourian et al., 2006) and 1.4 L/kg in infants and children (Ambrose, 1984), reflecting its lipophilic character and LogP of 1.14, with active concentrations achieved in the brain and CNS, various organs, and a range of body fluids, such as breast milk, prostatic fluid, saliva, aqueous and vitreous humour, and pleural fluid (Ambrose, 1984).
Both Gram positive and Gram negative bacterial cells effectively internalise and accumulate chloramphenicol to achieve a 1000-fold difference between external and internal concentrations; there is evidence that chloramphenicol traverses the outer membrane through a porin (pore protein) and crosses the inner membrane by active transport that is temperature-, pH-, and medium-dependent (Danilchanka et al., 2008). For more on the bioavailability of chloramphenicol, see Powell and Nahata (1982) and Ambrose (1984).
4.4.4 Mode of Action and Selectivity
Chloramphenicol inhibits protein synthesis through binding to the large ribosome subunit (50S) at the peptidyl transferase centre A site, thus preventing binding of the next charged tRNA (Schlünzen et al., 2001; Davidovitch et al., 2008), and it has been suggested that the binding of chloramphenicol to this site is a result of its similarity in structure to an aminoacylated nucleoside (Hansen et al., 2003). Chloramphenicol has a number of structural features that facilitate this binding through hydrogen bonding: the oxygens of the nitro group (to C2452 and U2504), the 1-OH group (to G2061), the carbonyl group (to U2506), and the 3-OH group, which is crucial for antibacterial activity (to U2506 and G2484, as well as interacting with a Mg2+ ion at the catalytic site).
The selectivity of chloramphenicol binding has been suggested to arise due to differences between the conformations of bacterial and eukaryotic peptidyl transferase center (PTC), such that chloramphenicol only binds to the A site of the bacterial ribosome and so only inhibits bacterial protein synthesis (Yonath, 2005). Chloramphenicol is, however, known to inhibit mitochondrial protein synthesis to some extent and causes serious side effects when used systemically (see Subsection 4.4.6 for more detail).
4.4.5 Bacterial Resistance
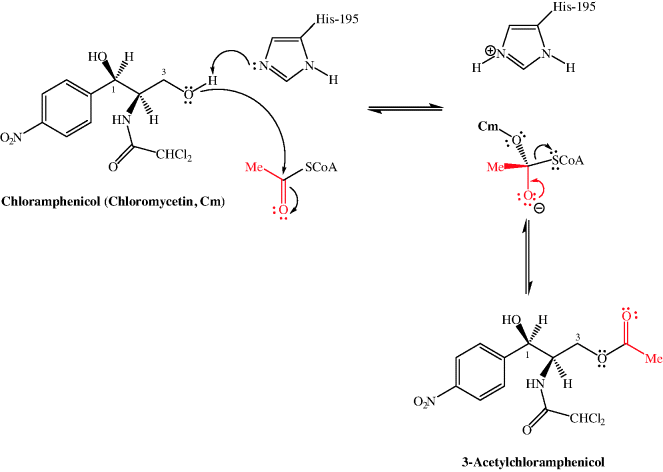
In Subsection 4.4.4, we learned that the 3-hydroxyl group of chloramphenicol was fundamental to its antibacterial activity, so it will not be a surprise to learn that one of the ways in which bacteria can develop resistance to chloramphenicol is through the chemical modification of this group. Resistance arises due to chloramphenicol acetyltransferases (CATs), which acetylate chloramphenicol so that it binds less effectively to the PTC A site (for example, there is a 20-fold decrease in the affinity of Escherichia coli ribosomes for the 3-acetylated product (Shaw and Unowsky, 1968)). CAT genes are both plasmid- (e.g. CatB2, Salmonella typhimurium) and chromosome-derived (e.g. CatB3, Pseudomonas aeruginosa) (Schwartz et al., 2004). The mechanism for the acetylation of chloramphenicol involves the deprotonation of the less-hindered primary hydroxyl group (3-OH) by the imidazole ring of histidine residue 195 of the active site (which is conserved in all CAT variants), followed by attack of the oxyanion generated upon acetyl CoA to give a tetrahedral intermediate and the loss of the CoA thiolate anion to give the acetylated product (Scheme 4.4.2) (Shaw, 1991).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


