Chemotherapy of Tuberculosis, Mycobacterium Avium Complex Disease, and Leprosy
Mycobacteria cause tuberculosis (TB) and leprosy. Although the burden of leprosy has decreased, TB is still the most important infectious killer of humans. Mycobacterium avium-intracellulare (or Mycobacterium avium complex [MAC]) infection continues to be difficult to treat, mainly due to 3 natural barriers:
• Cell wall—More than 60% of the cell wall is lipid, mainly mycolic acids composed of 2-branched, 73-hydroxy fatty acids with chains made of 76-90 carbon atoms. This shield prevents many pharmacological compounds from getting to the bacterial cell membrane or inside the cytosol.
• Efflux pumps—These transport proteins export potentially harmful chemicals from the bacterial cytoplasm into the extracellular space, preventing accumulation of effective drug concentrations in the cell. These exporters are responsible for the native resistance of mycobacteria to many standard antibiotics. As an example, ATP-binding cassette (ABC) permeases comprise a full 2.5% of the genome of Mycobacterium tuberculosis.
• Location in host—Some of the bacilli hide inside the patient’s cells, adding an extra physicochemical barrier that antimicrobial agents must cross to be effective.
Mycobacteria are defined by their rate of growth on agar as rapid and slow growers (Table 56–1). Slow growers tend to be susceptible to antibiotics developed specifically for Mycobacteria; rapid growers tend to be also susceptible to antibiotics used against many other bacteria.
Table 56–1
Pathogenic Mycobacterial Rapid and Slow Growers (Runyon Classification)

The mechanisms of action of the anti-mycobacterial drugs are summarized in Figure 56–1. Figure 56–2 depicts the mechanisms of resistance to these drugs. Tables 56–2 and 56–3 summarize the pharmacokinetic parameters of the antimycobacterial agents.
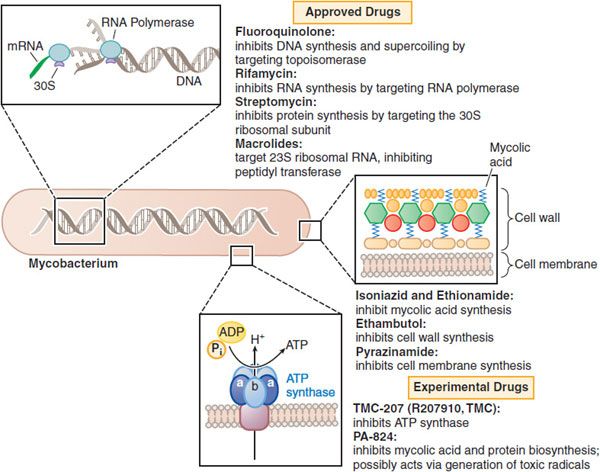
Figure 56–1 Mechanisms of action of established and experimental drugs used for the chemotherapy of mycobacterial infections. Shown at the top are the sites of action of approved drugs for the chemotherapy of mycobacterial diseases. Rifamycin is a generic term for several drugs, of which rifampin is used most frequently. Also included are 2 experimental drugs now under investigation: TMC-207 and PA-824. Clofazimine, whose mode of action is not understood, is omitted.
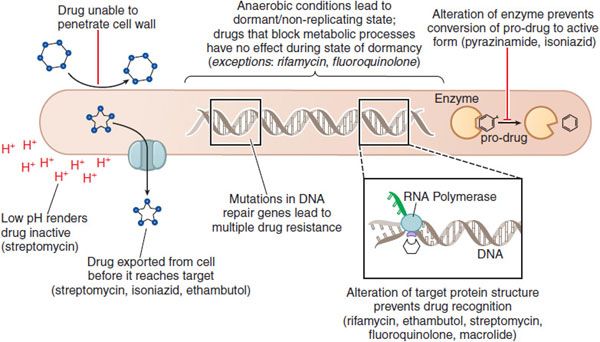
Figure 56–2 Mechanisms of drug resistance in Mycobacteria.
ANTI-MYCOBACTERIAL DRUGS
RIFAMYCINS: RIFAMPIN, RIFAPENTINE, AND RIFABUTIN
Rifampin or rifampicin (RIFADIN; RIMACTANE, others), rifapentine (PRIFTIN), and rifabutin (MYCOBUTIN) are important in treatment of mycobacterial diseases.
MECHANISM OF ACTION. Rifampin’s action against M. tuberculosis typifies the mechanism by which rifamycins act. Rifampin enters bacilli in a concentration-dependent manner, achieving steady-state concentrations within 15 min. The drug binds to the β subunit of DNA-dependent RNA polymerase (rpoB) to form a stable drug–enzyme complex, suppressing chain formation in RNA synthesis.
ANTIBACTERIAL ACTIVITY. Rifampin inhibits the growth of most gram-positive bacteria as well as many gram-negative microorganisms. Rifampin inhibits the growth of many M. tuberculosis clinical isolates in vitro at concentrations of 0.06-0.25 mg/L. Rifampin is also bactericidal against Mycobacterium leprae. Mycobacterium kansasii is inhibited by 0.25-1 mg/L. Most strains of Mycobacterium scrofulaceum, Mycobacterium intracellulare, and M. avium are suppressed by concentrations of 4 mg/L. Mycobacterium fortuitum is highly resistant to the drug. Rifabutin inhibits the growth of most MAC isolates at concentrations ranging from 0.25-1 mg/L and of many strains of M. tuberculosis at concentrations of ≤0.125 mg/L.
BACTERIAL RESISTANCE. The prevalence of rifampin-resistant isolates (1 in every 107 to 108 bacilli) is due to altered rpoB. Mutations in genes involved in DNA repair mechanisms will impair the repair of multiple genes, which may lead to hyper-mutable strains (see Chapter 48). M. tuberculosis Beijing genotype clinical isolates have been associated with higher rates of simultaneous rifampin and isoniazid resistance associated with mutations in the repair genes mut and ogt. Inducible or environment-dependent mutators may be more common than these stable mutator phenotypes. Antibiotics, endogenous oxidative and metabolic stressors lead to DNA damage, which induces dnaE2. The induction is associated with error-prone DNA repair and leads to higher rates of rifampin resistance.
ADME. After oral administration, the rifamycins are absorbed to variable extents (Table 56–2). Food decreases the rifampin CPmax by one-third; a high-fat meal increases the area under the curve (AUC) of rifapentine by 50%. Food has no effect on rifabutin absorption. Rifapentine should be taken with food, if possible. Rifamycins are metabolized by microsomal B-esterases and cholinesterases. A major pathway for rifabutin elimination is CYP3A. Due to autoinduction, all 3 rifamycins reduce their own AUCs with repeated administration (Table 56–3). They have good penetration into many tissues, but levels in the CNS reach only ~5% of those in plasma, likely due to the activity of P-glycoprotein. The drugs and metabolites are excreted by bile and eliminated via feces, with urine elimination accounting for only one-third and less of metabolites. Tables 56–2 and 56–3 summarize the pharmacokinetic parameters of the rifamycins.
Table 56–2
Population Pharmacokinetic Parameter Estimates for Antimycobacterial Drugs in Adult Patients
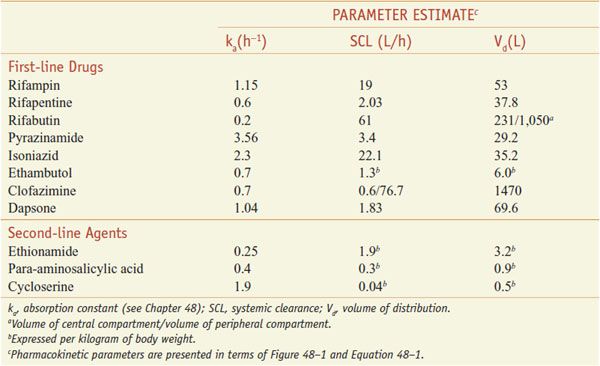
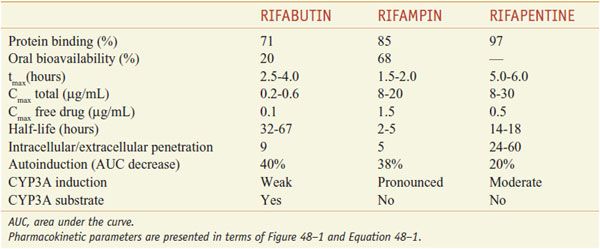
Table 56–3
Pharmacokinetic Parameters of Rifampin, Rifabutin, and Rifapentine
PHARMACOKINETICS-PHARMACODYNAMICS. Rifampin’s bactericidal activity is best optimized by a high AUC/MIC ratio. However, resistance suppression and rifampin’s enduring post-antibiotic effect are best optimized by high Cmax/MIC. Therefore, the duration of time that the rifampin concentration persists above the MIC is of less importance. These results predict that the t1/2 of a rifamycin is less of an issue in optimizing therapy, and that if patients could tolerate it, higher doses would lead to higher bactericidal activities while suppressing resistance.
THERAPEUTIC USES. Rifampin for oral administration is available alone or as a fixed-dose combination with isoniazid (150 mg of isoniazid, 300 mg of rifampin; Rifamate, others) or with isoniazid and pyrazinamide (50 mg of isoniazid, 120 mg of rifampin, and 300 mg pyrazinamide; RIFATER). A parenteral form of rifampin is also available. The dose of rifampin for treatment of TB in adults is 600 mg, given once daily, either 1 h before or 2 h after a meal. Children should receive 10-20 mg/kg given in the same way. Rifabutin is administered at 5 mg/kg/day and rifapentine at 10 mg/kg once a week. Rifampin is also useful for the prophylaxis of meningococcal disease and Haemophilus influenzae meningitis. To prevent meningococcal disease, adults may be treated with 600 mg twice daily for 2 days or 600 mg once daily for 4 days; children >1 month of age should receive 10-15 mg/kg, to a maximum of 600 mg. Combined with a β-lactam antibiotic or vancomycin, rifampin may be useful for therapy in selected cases of staphylococcal endocarditis or osteomyelitis.
UNTOWARD EFFECTS. Rifampin is generally well tolerated in patients. Fewer than 4% of patients with TB develop significant adverse reactions; the most common are rash, fever, and nausea and vomiting. Chronic liver disease, alcoholism, and old age appear to increase the incidence of severe hepatic problems. GI disturbances have occasionally required discontinuation of the drug. Various nonspecific symptoms related to the nervous system also have been noted.
Hypersensitivity reactions may be encountered. Adverse events associated with high rifampin dose are more commonly encountered when the time between doses is long; thus, high-dose rifampin should not be administered on a dosing schedule of less than twice weekly; less frequent administration is associated with a flu-like syndrome of fever, chills, and myalgias in 20% of patients; adverse effects may also include eosinophilia, interstitial nephritis, acute tubular necrosis, thrombocytopenia, hemolytic anemia, and shock. Light chain proteinuria, thrombocytopenia, transient leukopenia, and anemia have occurred during rifampin therapy. Rifampin crosses the placenta and its teratogenic potential; thus, its use is best avoided during pregnancy.
Rifabutin is generally well tolerated; primary reasons for discontinuation of therapy include rash (4%), GI intolerance (3%), and neutropenia (2%; 25% in patients with severe HIV infection). Uveitis and arthralgias have occurred in patients receiving rifabutin doses >450 mg daily in combination with clarithromycin or fluconazole. Patients should be cautioned to discontinue the drug if visual symptoms (pain or blurred vision) occur. Rifabutin causes an orange-tan discoloration of skin, urine, feces, saliva, tears, and contact lenses, like rifampin. Rarely, thrombocytopenia, a flu-like syndrome, chest pain, and hepatitis develop in patients treated with rifabutin. Unique side effects include polymyalgia, pseudojaundice, and anterior uveitis.
RIFAMYCIN OVERDOSE. Rifampin overdose is uncommon. The most prominent symptoms are the orange discoloration of skin, fluids, and mucosal surfaces, leading to the term red-man syndrome. Overdose can be life-threatening; treatment consists of supportive measures; there is no antidote.
DRUG INTERACTIONS. Because rifampin potently induces CYPs, its administration results in a decreased t1/2 for a number of compounds that are metabolized by these CYPs. Rifabutin is a less potent inducer of CYPs than rifampin; however, rifabutin does induce hepatic microsomal enzymes and decreases the t1/2 of zidovudine, prednisone, digitoxin, quinidine, ketoconazole, propranolol, phenytoin, sulfonylureas, and warfarin. It has less effect than rifampin on serum levels of indinavir and nelfinavir. Compared to rifabutin and rifampin, the CYP-inducing effects of rifapentine are intermediate.
PYRAZINAMIDE
Pyrazinamide is the synthetic pyrazine analog of nicotinamide.
MECHANISM OF ACTION. Pyrazinamide is activated to pyrazinoic acid under acidic conditions that likely prevail at the edges of necrotic TB cavities where inflammatory cells produce lactic acid. Some of the parent drug diffuses into M. tuberculosis where a nicotinamidase (pyrazinaminidase) deaminates pyrazinamide to pyrazinoic acid (POA?), which is exported by an efflux pump. In an acidic extracellular milieu, a fraction of POA? is protonated to POAH and enters the bacillus. As the pH of the extracellular medium declines toward the pKa of pyrazinoic acid, 2.9, the Henderson-Hasselbalch equilibrium (see Figure 2–3) progressively favors the formation of POAH, its equilibration across bacillar membrane, and its accumulation within the bacillus; acidic conditions also enhance microbial killing. Although the actual mechanism of microbial kill is still unclear, 4 mechanisms have been proposed:
• Inhibition of fatty acid synthase type I leading to interference with mycolic acid synthesis
• Binding to ribosomal protein S1 (RpsA) and inhibition of trans-translation
• Reduction of intracellular pH disruption of membrane transport by HPOA
ANTIBACTERIAL ACTIVITY. Pyrazinamide exhibits antimicrobial activity in vitro only at acidic pH. At pH of 5.8-5.95, 80-90% of clinical isolates have an MIC of ≤100 mg/L.
MECHANISMS OF RESISTANCE. Pyrazinamide-resistant M. tuberculosis express nicotinamidase with reduced affinity for pyrazinamide. This reduced affinity decreases the conversion of pyrazinamide to POA. Single point mutations in the pncA gene are encountered in up to 70% of resistant clinical isolates.
ADME. Oral bioavailability of pyrazinamide is >90%. GI absorption segregates patients into 2 groups: fast absorbers (56%) with an absorption rate constant of 3.56/h and slow absorbers (44%) with an absorption rate of 1.25/h. The drug is concentrated 20-fold in lung epithelial lining fluid. Pyrazinamide is metabolized by microsomal deamidase to POA and subsequently hydroxylated to 5-hydroxy-POA, which is excreted by the kidneys. CL (clearance) and Vd (volume of distribution) increase with patient mass (0.5 L/h and 4.3 L for every 10 kg above 50 kg), and Vd is larger in males (by 4.5 L) (see Table 56–2). This has several implications: the t1/2 of pyrazinamide will vary considerably based on weight and gender, and the AUC0-24 will decrease with increase in weight for the same dose (same mg drug/kg body weight). Pyrazinamide clearance is reduced in renal failure; therefore, the dosing frequency must be reduced to 3 times a week at low glomerular filtration rates. Hemodialysis removes pyrazinamide; therefore, the drug needs to be re-dosed after each session of hemodialysis.
MICROBIAL PHARMACOKINETICS-PHARMACODYNAMICS. Pyrazinamide’s sterilizing effect is closely linked to AUC0-24/MIC. However, resistance suppression is linked to the fraction of time that CP persists above MIC (T > MIC). Because patient weight impacts both SCL and volume, both AUC and t1/2 will be impacted by high weight. Simulations indicate that optimal AUC0-24/MIC and T > MIC are likely to be achieved only by doses much higher than the currently recommended 15-30 mg/kg/day; the safety of such higher doses in patients is unclear.
THERAPEUTIC USES. The coadministration of pyrazinamide with isoniazid or rifampin has led to a one-third reduction in the duration of anti-TB therapy, and a two-thirds reduction in TB relapse. The combination has reduced the length of therapy to 6 months, producing the current “short course” regimen. Pyrazinamide is administered at an oral dose of 15-30 mg/kg/day.
UNTOWARD EFFECTS. Injury to the liver is the most serious side effect of pyrazinamide. With an oral dose of 40-50 mg/kg, signs and symptoms of hepatic disease appear in ~15% of patients, with jaundice in 2-3% and death due to hepatic necrosis in rare instances. Current regimens (15-30 mg/kg/day) are much safer. Prior to pyrazinamide administration, all patients should undergo studies of hepatic function, and these studies should be repeated at frequent intervals during the entire period of treatment. If there is evidence of significant hepatic damage, therapy must be stopped. Pyrazinamide should not be given to patients with hepatic dysfunction unless its use is unavoidable.
In nearly all patients, pyrazinamide inhibits excretion of urate, and causes a hyperuricemia that may result in acute episodes of gout. Other untoward effects observed with pyrazinamide include arthralgias, anorexia, nausea and vomiting, dysuria, malaise, and fever. Pyrazinamide is not approved in the U.S. during pregnancy because of inadequate data on teratogenicity.
ISONIAZID
Isoniazid (isonicotinic acid hydrazide, INH; NYDRAZID, others) is a primary agent for TB. All patients infected with isoniazid-sensitive strains should receive the drug if they can tolerate it. The combination of isoniazid + pyrazinamide + rifampin provides a “short-course” therapy with improved remission rates.
MECHANISM OF ACTION. Isoniazid enters bacilli by passive diffusion. The drug is not directly toxic to the bacillus but must be activated to its toxic form within the bacillus by KatG, a multifunctional catalase-peroxidase. The activated drug forms adducts with bacillar NAD+ and NADP+ that inhibit essential steps in mycolic acid synthesis (cell wall) and nucleic acid synthesis (Figure 56–3). Other products of KatG activation of INH include superoxide, H2O2, alkyl hydroperoxides, and the NO radical, which may also contribute to the mycobactericidal effects of INH.
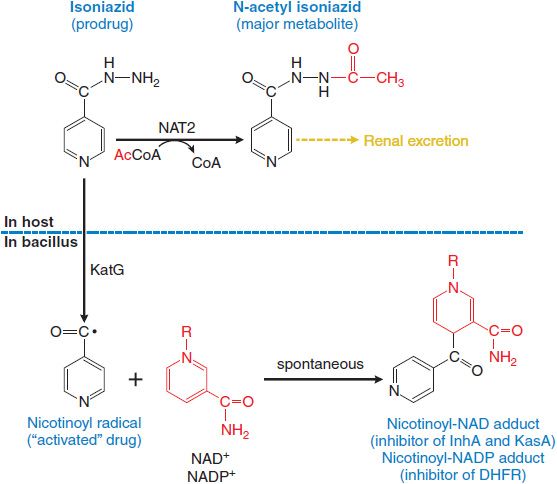
Figure 56–3 Metabolism and activation of isoniazid. metabolized in humans by NAT2 isoforms to its principal metabolite, N-acetyl isoniazid, which is excreted by the kidney. Isoniazid diffuses into mycoplasma where it is “activated” by KatG (oxidase/peroxidase) to the nicotinoyl radical. The nicotinoyl radical reacts spontaneously with NAD+ to produce adducts that inhibit essential enzymes in synthesis of the cell-wall component mycolic acid, and with NADP+ to produce an inhibitor of nucleic acid synthesis. InhA, enoyl acyl carrier protein; KasA, enoyl acyl carrier protein synthase; DHFR, dihydrofolate reductase.
MECHANISMS OF RESISTANCE. Resistance to INH is associated with mutation or deletion of katG, overexpression of the genes for inhA (confers low-level resistance to INH and some cross-resistance to ethionamide) and ahpC, and mutations in the kasA gene. The prevalence of drug-resistant mutants is ~1 in 106 bacilli. Because TB cavities may contain as many as 107-109 microorganisms, preexistent resistance can be expected in pulmonary TB cavities of untreated patients. These spontaneous mutants will be selected and amplified by monotherapy. Thus, 2 or more agents are usually used. Because the mutations resulting in drug resistance are independent events, the probability of resistance to 2 antimycobacterial agents is small, ~1 in 1012 (1 × 106 × 106), a low probability considering the number of bacilli involved.
ADME. The bioavailability of orally administered isoniazid is ~100% for the 300 mg dose. The pharmacokinetics of isoniazid are best described by a one-compartment model, with the pharmacokinetic parameters in Table 56–2. Isoniazid is metabolized by hepatic arylamine N-acetyltransferase type 2 (NAT2), encoded by a variety of NAT2* alleles (see Figure 56–3). Isoniazid clearance in patients has been traditionally classified as 1 of 2 phenotypic groups: “slow” and “fast” acetylators (Figure 56–4), and more recently as 3 groups, including “intermediate” metabolizers; this variability largely reflects expression of various NAT2 alleles. Most (75-95%) of a dose of isoniazid is excreted in the urine within 24 h, predominantly as acetylisoniazid and isonicotinic acid.
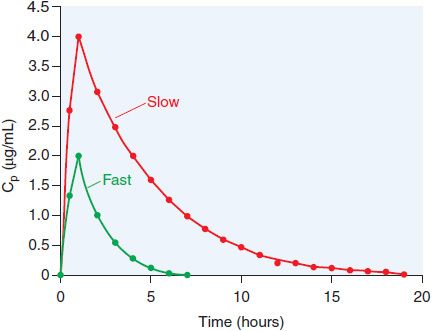
Figure 56–4 Multi-modal distribution of INH clearance due to NAT2 polymorphisms. A group of matched male volunteers received INH (250 mg orally) and the time courses of plasma drug levels (Cp) were assessed. One-third of the subjects had INH elimination t1/2 values >1.5 h; these are the fast acetylators
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


