Chemotherapy of Malaria
Malaria affects about a quarter of a billion people and leads to almost 900,000 deaths annually. This disease is caused by infection with single-celled protozoan parasites of the genus Plasmodium. Five Plasmodium spp. are known to infect humans: P. falciparum, P. vivax, P. ovale, P. malariae, and P. knowlesi. P. falciparum and P. vivax cause most of the malarial infections worldwide. P. falciparum accounts for the majority of the burden of malaria in sub-Saharan Africa and is associated with the most severe disease. P. vivax accounts for half of the malaria burden in South and East Asia and 80% of the malarial infections in the America.
BIOLOGY OF MALARIAL INFECTION
Plasmodium sporozoites, which initiate infection in humans, are inoculated into the dermis and enter the bloodstream following the bite of a Plasmodium-infected female anopheline mosquito. Within minutes, sporozoites travel to the liver, where they infect hepatocytes via cell surface receptor-mediated events. This process initiates the asymptomatic prepatent period, or exoerythrocytic stage of infection, which typically lasts 1 week.
During this period, the parasite undergoes asexual replication within hepatocytes, resulting in production of liver stage schizonts. When the infected hepatocytes rupture, tens of thousands of merozoites are released into the bloodstream and infect red blood cells. After the initial exoerythrocytic stage, P. falciparum and P. malariae are no longer found in the liver. P. vivax and P. ovale, however, can maintain a quiescent hepatocyte infection as a dormant form of the parasite known as the hypnozoite and can reinitiate symptomatic disease long after the initial symptoms of malaria are recognized and treated. Erythrocytic forms cannot reestablish infection of hepatocytes. Transmission of human-infecting malarial parasites is maintained in human populations by the persistence of hypnozoites (several months to few years for P. vivax and P. ovale), by antigenic variation in P. falciparum (probably months), and by the putative antigen variation in P. malariae (for as long as several decades).
The asexual erythrocytic stages of malarial parasites are responsible for the clinical manifestations of malaria. This part of the Plasmodium life cycle is initiated by merozoite recognition of red blood cells, mediated by cell surface receptors, followed by red blood cell invasion.
Once inside a red blood cell, the merozoite develops into a ring form, which becomes a trophozoite that matures into an asexually dividing blood stage schizont. Upon rupture of the infected erythrocyte, these schizonts release 8-32 merozoites that can establish new infections in nearby red blood cells. The erythrocytic replication cycle lasts for 24 h (for P. knowlesi), 48 h (for P. falciparum, P. vivax, and P. ovale), and 72 h (for P. malariae). Although most invading merozoites develop into schizonts, a small proportion become gametocytes, the form of the parasite that is infective to mosquitoes. Gametocytes are ingested into the mosquito midgut during an infectious blood meal and then transform into gametes that can fertilize to become zygotes. Zygotes mature into ookinetes, which penetrate the mosquito midgut wall and develop into oocysts. Numerous rounds of asexual replication occur in the oocyst to generate sporozoites over 10-14 days. Fully developed sporozoites rupture from oocysts and invade the mosquito salivary glands, from which they can initiate a new infection during subsequent mosquito blood meals (Figure 49–1).
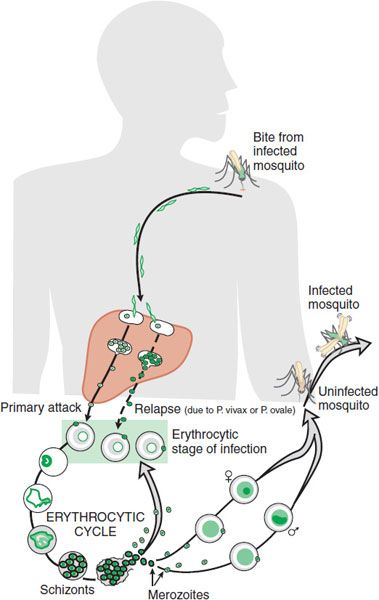
Figure 49–1 Life cycle of malaria parasites.
P. falciparum has a family of binding proteins that can recognize a variety of host cell molecules; it invades all stages of erythrocytes and therefore can achieve high parasitemias. P. vivax selectively binds to the Duffy chemokine receptor protein as well as reticulocyte-specific proteins. P. falciparum assembles cytoadherence proteins (PfEMP1s, encoded by a highly variable family of var genes) into structures called knobs that are presented on the erythrocyte surface. Knobs allow the P. falciparum-parasitized erythrocyte to bind to postcapillary vascular endothelium, so as to avoid spleen-mediated clearance and allow the parasite to grow in a low O2, high CO2 microenvironment.
The cardinal signs and symptoms of malaria are high, spiking fevers, chills, headaches, myalgias, malaise, and GI symptoms. The clinical manifestations of malaria are detailed in Chapter 49 of the 12th edition of the parent text.
CLASSIFICATION OF ANTIMALARIAL AGENTS
The various stages of the malarial parasite life cycle in humans differ in their drug sensitivity. Thus, antimalarial drugs can be classified based on their activities during this life cycle as well as by their intended use for either chemoprophylaxis or treatment. The spectrum of antimalarial drug activity leads to several generalizations.
The first relates to chemoprophylaxis: because no antimalarial drug kills sporozoites, it is not truly possible to prevent infection; drugs can only prevent the development of symptomatic malaria caused by the asexual erythrocytic forms.
The second relates to the treatment of an established infection: no single antimalarial is effective against all liver and intraerythrocytic stages of the life cycle that may coexist in the same patient. Complete elimination of the parasite infection, therefore, may require more than 1 drug.
The patterns of clinically useful activity fall into 3 general categories.
The first group of agents (artemisinins, chloroquine, mefloquine, quinine and quinidine, pyrimethamine, sulfadoxine, and tetracycline) are not reliably effective against primary or latent liver stages. Instead, their action is directed against the asexual blood stages responsible for disease. These drugs will treat, or prevent, clinically symptomatic malaria.
The second group of drugs (typified by atovaquone and proguanil) target not only the asexual erythrocytic forms but also the primary liver stages of P. falciparum. This additional activity shortens to several days the required period for postexposure chemoprophylaxis.
The third category, comprised solely of primaquine, is effective against primary and latent liver stages as well as gametocytes. Primaquine is used most commonly to eradicate the intrahepatic hypnozoites of P. vivax and P. ovale that are responsible for relapsing infections.
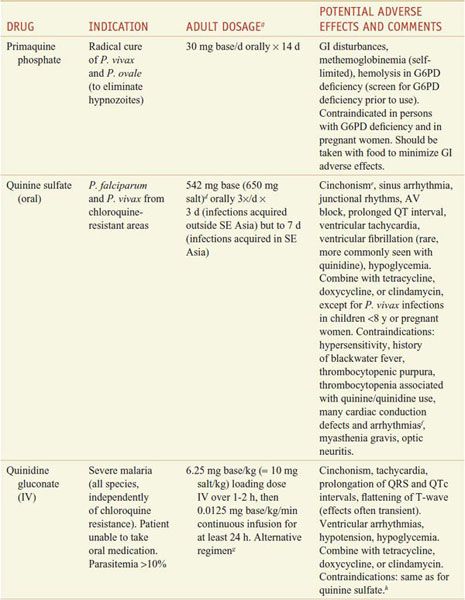
Aside from their antiparasitic activity, the utility of antimalarials for chemoprophylaxis or therapy depends on their pharmacokinetics and their safety. Quinine and primaquine, which have significant toxicity and relatively short half-lives, generally are reserved for the treatment of established infection and are not used for chemoprophylaxis in a healthy traveler. By contrast, chloroquine is relatively free from toxicity and has a long t1/2 that is convenient for chemoprophylactic dosing (in those few areas still reporting chloroquine-sensitive malaria) (see Tables 49–1 and 49–2).
Table 49–1
Regimens for the Prevention of Malaria in Non-immune Adults
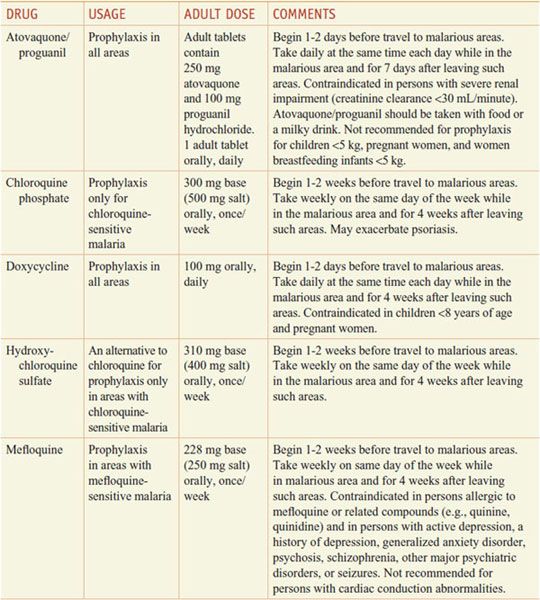
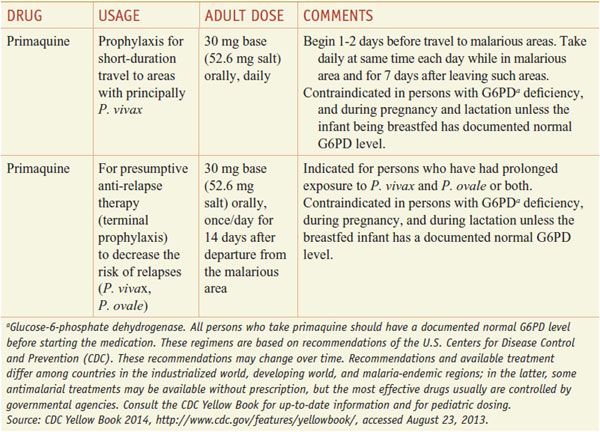
Table 49–2
Regimens for the Treatment of Malaria
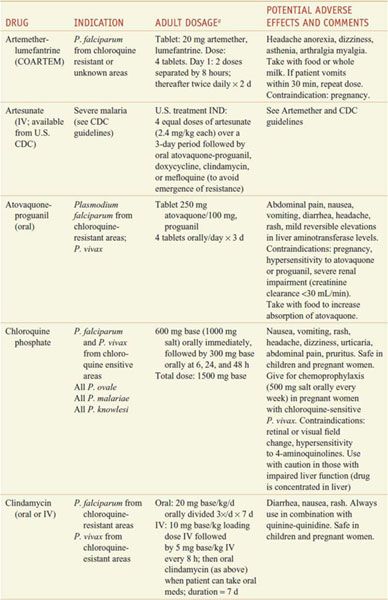


For ease of reference, detailed information on the antimalarial drugs appears below in alphabetical order by drug name.
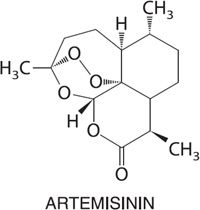
ARTEMISININ AND DERIVATIVES
Artemisinin and its 3 major semisynthetic derivatives in clinical use, dihydroartemisinin, artemether, and artesunate, are potent and fast-acting antimalarials. They are particularly well suited for the treatment of severe P. falciparum malaria and are also effective against the asexual erythrocytic stages of P. vivax. Increasingly, the standard treatment of malaria employs artemisinin-based combination therapies (ACTs) to increase treatment efficacy and reduce selection pressure for the emergence of drug resistance.
Artemisinins cause a significant reduction of the parasite burden, with a 4-log10 reduction in the parasite population for each 48-h cycle of intraerythrocytic invasion, replication, and egress. Only 3 to 4 cycles (6-8 days) of treatment are required to remove all the parasites from the blood. Additionally, artemisinins possess some gametocytocidal activity, leading to a decrease in malarial parasite transmission.
Mechanism of Action. The activity of artemisinin and derivatives seems to result from cleavage of the drug’s peroxide bridge by reduced heme-iron, produced inside the highly acidic digestive vacuole (DV) of the parasite as it digests hemoglobin. The site of action of the putatively toxic heme-adducts is unclear. Additionally, activated artemisinin might in turn generate free radicals that alkylate and oxidize macromolecules in the parasite.
ADME. The semisynthetic artemisinins have been formulated for oral (dihydroartemisinin, artesunate, and artemether), intramuscular (artesunate and artemether), intravenous (artesunate), and rectal (artesunate) routes. Bioavailability after oral dosing typically is <30%. Peak serum levels occur rapidly with artemisinins and in 2-6 h with intramuscular artemether. Both artesunate and artemether have modest levels of Asexual malarial parasites flourish plasma protein binding, ranging from 43-82%. These derivatives are extensively metabolized and converted to dihydroartemisinin, which has a plasma t1/2 of 1-2 h. Drug bioavailability via rectal administration is highly variable among individual patients. With repeated dosing, artemisinin and artesunate induce their own CYP-mediated metabolism, primarily via CYPs 2B6 and 3A4. This may enhance clearance by up to 5-fold.
Therapeutic Uses. Given their rapid and potent activity against even multidrug-resistant parasites, the artemisinins are valuable for the treatment of severe P. falciparum malaria. The artemisinins generally are not used alone because of their limited ability to eradicate infection completely. Artemisinins are highly effective, when combined with other antimalarials, for the first-line treatment of malaria. Artemisinins should not be used for chemoprophylaxis because of their short t1/2.
Toxicity and Contraindications. In pregnant rats and rabbits, artemisinins can cause increased embryo lethality or malformations early postconception. Preclinical toxicity studies have identified the brain (and brainstem), liver, bone marrow, and fetus as the principal target organs. However, no systematic neurological changes were attributable to treatment in patients >5 years of age. Patients may develop dose-related and reversible decreases in reticulocyte and neutrophil counts and increases in transaminase levels. About 1 in 3000 patients develops an allergic reaction. Studies of artemisinin treatment during the first trimester have found no evidence of adverse effects on fetal development. Nonetheless, it is recommended that ACTs not be used for the treatment of children <5 kg or during the first trimester of pregnancy.
ACT PARTNER DRUGS. Current ACT regimens that are well tolerated in adults and children ≥5 kg include artemether/lumefantrine, artesunate-mefloquine, artesunate-amodiaquine, artesunate-sulfadoxine-pyrimethamine, and dihydroartemisinin-piperaquine.
Lumefantrine shares structural similarities with the arylamino alcohol drugs mefloquine and halofantrine and is formulated with artemether (COARTEM). This combination is highly effective for the treatment of uncomplicated malaria and is the most widely used first-line antimalarial across Africa. The pharmacokinetic properties of lumefantrine include a large apparent volume of distribution and a terminal elimination t1/2 of 4-5 days. Administration with a high-fat meal is recommended because it significantly increases absorption. A sweetened dispersible formulation of artemether-lumefantrine (COARTEM DISPERSIBLE) has been approved for treatment of children.
Amodiaquine is a congener of chloroquine that is no longer recommended in the U.S. for chemoprophylaxis of P. falciparum malaria because of its toxicity (hepatic and agranulocytosis) that were generally associated with its prophylactic use. Amodiaquine is rapidly converted by hepatic CYPs into monodesethyl-amodiaquine. This metabolite, which retains substantial antimalarial activity, has a plasma t1/2 of 9-18 days and reaches a peak concentration of ~500 nM 2 h after oral administration. By contrast, amodiaquine has a t1/2 of ~3 h, attaining a peak concentration of ~25 nM within 30 min of oral administration. Clearance rates of amodiaquine ranges between individuals from 78-943 mL/min/kg.
Piperaquine is a potent and well-tolerated bisquinoline compound structurally related to chloroquine. Piperaquine has a large volume of distribution and reduced rates of excretion after multiple doses. It is rapidly absorbed, with a Tmax (time to reach the highest concentration) of 2 h after a single dose. Piperaquine has the longest plasma t1/2 (5 weeks) of all ACT partner drugs, which might also be effective in reducing rates of reinfection following treatment.
Pyronaridine is an antimalarial structurally related to amodiaquine. It is well tolerated and highly potent against both P. falciparum and P. vivax, causing fever to subside in 1-2 days and parasite clearance in 2-3 days.
ATOVAQUONE
A fixed combination of atovaquone with proguanil hydrochloride (MALARONE) is available in the U.S. for malaria chemoprophylaxis and for the treatment of uncomplicated P. falciparum malaria in adults and children.
Antimalarial Action and Resistance. Atovaquone is a lipophilic analog of ubiquinone, the electron acceptor for parasite dihydroorotate dehydrogenase, an enzyme essential for pyrimidine biosynthesis in the parasite. Atovaquone inhibits electron transport, collapses the mitochondrial membrane potential, and inhibits regeneration of ubiquinone. The drug is highly active against P. falciparum asexual blood stage parasites and the liver stages of P. falciparum, but not against P. vivax liver stage hypnozoites. Synergy between proguanil and atovaquone results from the ability of nonmetabolized proguanil to enhance the mitochondrial toxicity of atovaquone. Resistance to atovaquone alone in P. falciparum develops easily and is conferred by single nonsynonymous nucleotide polymorphisms in the cytochrome b gene located in the mitochondrial genome. Addition of proguanil markedly reduces the frequency of appearance of atovaquone resistance. However, once atovaquone resistance is present, the synergy of the partner drug proguanil diminishes.
ADME. Atovaquone absorption is slow and variable after an oral dose due to its lipophilicity. Absorption improves when the drug is taken with a fatty meal. More than 99% of the drug is bound to plasma protein; cerebrospinal fluid levels are <1% of those in plasma. Profiles of drug concentration versus time often show a double peak; the first at 1-8 h, the second 1-4 days after a single dose; this pattern suggests an enterohepatic circulation. In the absence of a CYP-inducing second medication, humans do not metabolize atovaquone significantly. The drug is excreted in bile, and >94% of the drug is recovered unchanged in feces. Atovaquone has a reported elimination t1/2 from plasma of 2-3 days in adults and 1-2 days in children.
Therapeutic Uses. A tablet containing a fixed dose of 250 mg atovaquone and 100 mg proguanil hydrochloride, taken orally, is highly effective and safe in a 3-day regimen for treating mild-to-moderate attacks of chloroquine- or sulfadoxine-pyrimethamine-resistant P. falciparum malaria. The same regimen followed by a primaquine course is effective in treatment of P. vivax malaria. Atovaquone-proguanil is a standard agent for malaria chemoprophylaxis. Experience in prevention of non–P. falciparum malaria is limited. P. vivax infection may occur after drug discontinuation, indicating imperfect activity against exoerythrocytic stages of this parasite.
Toxicity. Atovaquone may cause side effects (abdominal pain, nausea, vomiting, diarrhea, headache, rash) that require cessation of therapy. Vomiting and diarrhea may decrease drug absorption, resulting in therapeutic failure. However, readministration of this drug within an hour of vomiting may still be effective in patients with P. falciparum malaria. Atovaquone occasionally causes transient elevations of serum transaminase or amylase.
Precautions and Contraindications. Although atovaquone is generally considered to be safe, it needs further evaluation in children <11 kg, pregnant women, and lactating mothers. Atovaquone may compete with certain drugs for binding to plasma proteins, and therapy with rifampin, a potent inducer of CYP-mediated drug metabolism, can reduce plasma levels of atovaquone substantially, whereas atovaquone may raise plasma levels of rifampin. Coadministration with tetracycline is associated with a 40% reduction in plasma concentration of atovaquone.
DIAMINOPYRIMIDINES
Sulfadoxine-pyrimethamine (FANSIDAR) was a primary treatment for uncomplicated P. falciparum malaria, especially against chloroquine-resistant strains. Due to widespread resistance, it is no longer recommended for the treatment of uncomplicated malaria.
Antimalarial Action and Resistance. Pyrimethamine is a slow-acting blood schizontocide with antimalarial effects in vivo resulting from inhibition of folate biosynthesis in Plasmodium, similar to proguanil. The efficacy of pyrimethamine against hepatic forms of P. falciparum is less than that of proguanil, and at therapeutic doses pyrimethamine fails to eradicate P. vivax hypnozoites or gametocytes of any Plasmodium species. It increases the number of circulating P. falciparum mature infecting gametocytes, likely leading to increased transmission to mosquitoes during the period of treatment.
Synergy of pyrimethamine and the sulfonamides or sulfones results from inhibition of 2 metabolic steps in folate biosynthesis in the parasite:
• The utilization of p-aminobenzoic acid for the synthesis of dihydropteroic acid, which is catalyzed by dihydropteroate synthase and inhibited by sulfonamides
• The reduction of dihydrofolate to tetrahydrofolate, which is catalyzed by dihydrofolate reductase and inhibited by pyrimethamine (see
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


