Type of tumor
Characteristics
1. Cystic lesions
Cystic lesions are the commonest orbital lesions seen in the pediatric age group
Dermoid cysts are benign developmental choristomas sequestered in fetal suture lines at the time of closure
Cysts containing squamous epithelium without dermal appendages are called epidermoid cysts
Choristomatous tumors containing derivatives of all 3 germinal layers are teratomas
Congenital cystic eyes and colobomatous cysts are rare developmental anomalies
Acquired cysts are less common than developmental cysts
Mucocele and mucopyocele originate from paranasal sinuses, while dacryocele originates from the lacrimal sac
Encephalocele or meningocele result from congenital bony defects that permit herniation of intracranial tissues
These are usually benign in nature
A. Developmental cysts
(a) Dermoid, epidermoid, and choristoma
(b) Teratoma
(c) Congenital cystic eye
(d) Colobomatous cyst
B. Acquired cysts
(a) Parasitic cysts: hydatid, cysticercus
(b) Epithelial cysts (appendage/implantation)
(c) Lacrimal duct cysts
(d) Aneurysmal bone cyst
(e) Optic nerve sheath meningocele
C. Developing from adjacent structures
(a) Mucocele/mucopyocele
(b) Dacryocele
(c) Encephalocele/meningocele
2. Vascular lesions
(a) Hemangioma (capillary/cavernous)
(b) Lymphangioma
(c) Orbital varix
(d) Arteriovenous malformation
(e) Organized hematoma (hematic cyst/cholesterol granuloma)
(f) Hemangiopericytoma
(g) Sturge-Weber syndrome
Hemangiomas are hamartomatous growths composed of proliferating capillary endothelial cells, whereas malformations are developmental anomalies of capillary, venous, arterial, or lymphatic vessels
Hemangiomas can progress or regress with age although vascular malformations remain relatively static, with growth of lesions correlating with growth of the child
Hemangiopericytoma is a benign tumor of pericytes
Sturge-Weber syndrome is a capillary malformation of leptomeninges with or without ocular or facial involvement
Most vascular tumors are usually benign in nature, but some of them like hemangiopericytoma can have a malignant course
3. Inflammatory lesions
(a) Preseptal and orbital cellulitis
(b) Idiopathic orbital inflammatory syndrome
(c) Other inflammatory diseases
Inflammatory lesions can be of infective or noninfective origin
Preseptal and orbital cellulitis are infective diseases of preseptal and post-septal orbital tissues, respectively
Idiopathic orbital inflammatory disease (orbital pseudotumor) is an inflammatory proptosis of childhood. It significantly differs from the adult form Bilaterality, episodic recurrence, and systemic manifestations like headache, nausea, vomiting, and lethargy are common features
Thyroid eye disease, the commonest inflammatory lesion of adults, rarely occurs in children
Inflammatory diseases are benign lesions
4. Histiocytic and lymphoproliferative lesions
(a) Langerhans cell histiocytosis (histiocytosis X, Hand-Schuller-Christian disease, and Letterer-Siwe disease)
(b) Non-Langerhans cell histiocytosis (juvenile xanthogranuloma)
(c) Leukemia and lymphoma
(d) Sinus histiocytosis
Histiocytic disorders are caused by abnormal accumulation of cells of the mononuclear phagocytic system (dendritic cells and macrophages) which are derived from the bone marrow stem cells
Leukemia (lymphocytic more than myelocytic) commonly involves the orbit in children
Lymphoma in children very rarely involves the orbit
Burkitt lymphoma is the most likely form to involve the orbit
These tumors can have a benign or malignant course
Lymphoma and leukemia are always malignant
5. Mesodermal tumors
(a) Fibroma/myofibromatosis
(b) Lipoma
(c) Leiomyoma
(d) Fibrous dysplasia
(e) Giant cell granuloma
(f) Sarcoma (osteosarcoma, leiomyosarcoma, fibrosarcoma, malignant fibrous histiocytoma, and alveolar soft part sarcoma)
(g) Rhabdomyosarcoma
Mesodermal tumors are tumors of the soft tissues and fibro-osseous tissues of the orbit
They can be benign or malignant
Rhabdomyosarcoma is the most common malignant tumor of the orbit in children
In general, mesenchymal tumors are classified into benign or malignant disease, depending upon histopathological features like number of mitotic figures, nuclear cytoplasmic ratio, invasion of adjacent structures, etc.
Sarcomas always have a malignant course
6. Neurogenic tumors
(a) Glioma
(b) Meningioma
(c) Neurofibroma
(d) Schwannoma
(e) Esthesioneuroblastoma
(f) Paraganglioma
(g) Melanotic neuroectodermal tumor of infancy
Glioma is the most important orbital tumor of neural origin in children. It is usually a low-grade astrocytoma
Approximately 20 % of gliomas are associated with neurofibromatosis 1
Plexiform neurofibroma is nearly always associated with neurofibromatosis 1
Meningioma and schwannoma are rare in children
Most of these tumors are benign, although paraganglioma can be benign or malignant
Esthesioneuroblastoma is a malignant tumor
7. Lacrimal gland tumors
(a) Pleomorphic adenoma (benign mixed tumor)
(b) Adenoid cystic carcinoma
(c) Adenocarcinoma (malignant mixed tumor)
Lacrimal gland tumors are less common in children as compared to adults
A malignant tumor of the lacrimal gland is more common than a benign lesion in childhood
Pleomorphic adenoma is a benign tumor, whereas adenoid cystic carcinoma and adenocarcinoma are malignant tumors
8. Metastatic tumors
(a) Neuroblastoma
(b) Ewing sarcoma
(c) Wilms tumor
(d) Rhabdomyosarcoma (rarely)
The orbit is the most common site of ocular metastasis in children
Neuroblastoma is the most frequent source of orbital metastasis in childhood
Other tumors that can metastasize to the orbit are Ewing sarcoma and Wilms tumor
These are always malignant in nature
9. Intraocular tumors with orbital spread
(a) Retinoblastoma
(b) Medulloepithelioma
These are intraocular tumors that can progress to orbital tumors in the advanced stage
Retinoblastoma is a malignant tumor
Medulloepithelioma can be either benign or malignant
12.1.1 Clinical Approach
A thorough clinical assessment including ophthalmological, general physical and systemic examination should be undertaken. Investigations include a complete hemogram in which total and differential leukocytic counts, peripheral blood smear, imaging of the orbit and the eye with B-scan ultrasonography, and CT scan/MRI are being examined. In cases with suspected malignancy, a metastatic workup should include a chest X-ray and ultrasonography of abdomen, kidney and renal function tests. Fine-needle aspiration cytology or incisional biopsy may be required to establish the clinical diagnosis. Other investigations in selected cases include a bone marrow aspiration/biopsy, lumbar puncture, bone scan/positron emission tomography (PET)/CT scan, and fine-needle aspiration cytology (FNAC) or biopsy of regional lymph node, if enlarged.
12.1.2 Retinoblastoma
Retinoblastoma is the most common primary intraocular tumor of infancy and childhood. It is a tumor of the embryonic neural retina. It has an incidence of 1 in every 20,000 live births. About 90 % cases are diagnosed by the age of 3–4 years and 98 % by 5 years. Bilateral disease is diagnosed earlier then unilateral disease. The burden of the disease is more in developing nations especially Latin America, Africa, and Asia including India. It contributes to 4 % of all pediatric cancers. Retinoblastoma is highly sensitive to chemotherapy, and survival rates in developed countries are greater than 90 %. There is marked disparity in the mortality associated with retinoblastoma between developed and developing countries. In the developing world, 40–70 % of children with retinoblastoma die as opposed to only 3–5 % in the developed counties. This is predominantly due to delayed diagnosis leading to advanced disease (extraocular disease) at presentation (Hurwitz et al. 2011 and Dimaras et al. 2012).
12.1.2.1 Genetics and Inheritance
The retinoblastoma gene (RB1), encoded on chromosome 13q14, was the first described tumor suppressor gene. Constitutional loss of one RB1 allele causes cancer predisposition, and loss of the second allele in a developing retinal cell leads to retinoblastoma.
Retinoblastoma can be sporadic or inherited. Sporadic tumors are unilateral and unifocal and occur at an older age, while inherited tumors occur at an earlier age and are often bilateral and multifocal. One-third of all cases have bilateral tumors. All cases with bilateral disease have germline mutations of RB1 and are heritable (Fig. 12.1). Only a small proportion of unilateral tumors are heritable. Alfred Knudson’s “two-hit” model of oncogenesis proposes that two mutational events are required for development of retinoblastoma. In inherited retinoblastoma, the first hit is the mutation in the RB1 inherited in the germline, and the second is acquired in the somatic retinal cell. In sporadic retinoblastoma, both mutations occur in the somatic retinal cell. Most cases of hereditary retinoblastoma have spontaneous new germline mutation, while their parents have both wild-type RB1 alleles. The risk of an offspring inheriting an RB1 mutation from a parent with germline mutation of RB1 is 50 % and 97 % of these offsprings with the inherited mutation will go on to develop retinoblastoma. A constitutional (germline) mutation of RB1 also causes an increased risk of a second cancer of the lung, soft tissue, bladder, skin, bone, and brain, and this risk is even higher when these patients are treated with radiation therapy for their retinoblastoma. A small proportion (10–15 %) of unilateral tumors are also hereditary.
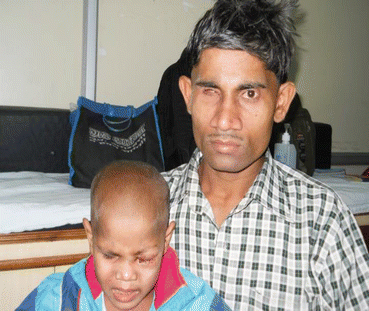
Fig. 12.1
Familial retinoblastoma
12.1.2.2 Clinical Presentation
Leukocoria (white pupillary reflex) is the most common presentation (Fig. 12.2). Retinoblastoma must be differentiated from Coat’s disease, cataract, toxocariasis, and retinopathy of prematurity which may also present with leukocoria. Pain may be present secondary to glaucoma; other presenting features are strabismus, redness of the eye, and poor vision. Orbital inflammation and fungating orbital mass are signs of advanced disease (Fig. 12.3). In developing countries, retinoblastoma presents very late in its extraocular stage, either with an orbital mass (proptosis) or with distant metastasis in the bone, bone marrow, lymph nodes, and central nervous system (Fig. 12.4a, b) Chawla et al. (2015).
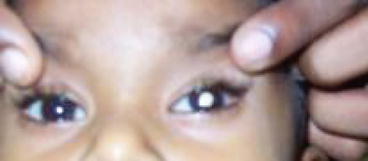
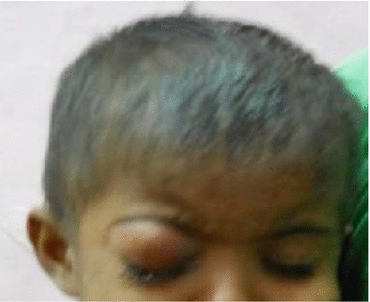
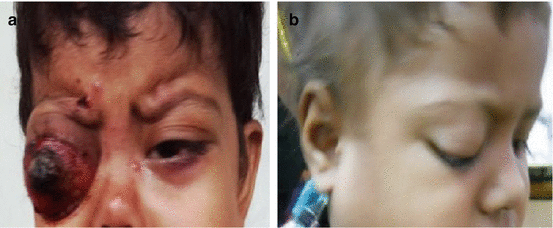
Fig. 12.2
Leukocoria
Fig. 12.3
Orbital cellulitis
Fig. 12.4
(a) Extraocular metastatic retinoblastoma. (b) RB with lymph node metastasis
12.1.2.3 Diagnosis
Diagnosis is established by characteristic ophthalmologic findings, often requiring examination under anesthesia. Imaging studies such as ultrasound, CT scan or MRI (preferable) scans are used for assessment of orbital, optic nerve, and intracranial extension (Fig. 12.5).
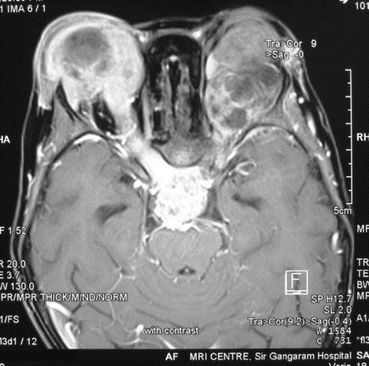
Fig. 12.5
Bilateral extraocular retinoblastoma with CNS metastasis
Rarely, children with hereditary retinoblastoma have pineal tumor (trilateral retinoblastoma) that may be found on imaging. CSF and bone marrow evaluation should only be done if indicated clinically, or by other imaging studies (i.e., in advanced disease).
Both eyes should be examined under general anesthesia.
Second malignant neoplasms are a major concern in retinoblastoma survival. Approximately 30 % of individuals cured of hereditary forms of RB will have a second malignancy within 30 years. Osteosarcoma is the commonest second malignancy; other second neoplasms include rhabdomyosarcoma and melanoma.
12.1.2.4 Classification and Staging
Intraocular
Intraocular retinoblastoma is localized to the eye and may be confined to the retina or may extend to involve other structures such as the choroid, ciliary body, anterior chamber, and the optic nerve head. Intraocular retinoblastoma does not extend beyond the eye into the tissues around the eye or to other parts of the body. For intraocular disease, enucleation is curative in more than 95 % of patients with advanced unilateral disease.
Extraocular
Extraocular retinoblastoma has extended beyond the eye. It may be confined to the tissues around the eye (orbital retinoblastoma), or it may have spread to the central nervous system, bone marrow, or lymph nodes (metastatic retinoblastoma).
Classification of Intraocular RB
Reese-Ellsworth Classification for Intraocular Tumors
Reese and Ellsworth developed a classification system for intraocular retinoblastoma that has been shown to have prognostic significance for maintenance of sight and control of local disease at a time when surgery and external beam radiation therapy (EBRT) were the primary treatment options:
Group I: very favorable for maintenance of sight
1.
Solitary tumor, smaller than 4 disk diameters (DD), at or behind the equator
2.
Multiple tumors, none larger than 4 DD, all at or behind the equator
Group II: favorable for maintenance of sight
1.
Solitary tumor, 4–10 DD at or behind the equator
2.
Multiple tumors, 4–10 DD, behind the equator
Group III: possible for maintenance of sight
1.
Any lesion anterior to the equator
2.
Solitary tumor, larger than 10 DD, behind the equator
Group IV: unfavorable for maintenance of sight
1.
Multiple tumors, some larger than 10 DD
2.
Any lesion extending anteriorly to the ora serrata
Group V: very unfavorable for maintenance of sight
1.
Massive tumors involving more than one half of the retina
2.
Vitreous seeding
International Classification of Intraocular Retinoblastoma
With the introduction of systemic chemotherapy as the primary modality for globe salvage in intra ocular disease, there is a new grouping system for retinoblastoma, which may offer greater precision in stratifying risk for newer therapies. The International Classification of Retinoblastoma that is used in the current Children’s Oncology Group treatment studies and in some institutional studies has been shown to assist in predicting those eyes that are likely to be cured without the need for enucleation or external beam radiotherapy.
12.1.2.5 Staging
The patients of are usually diagnosed upon ophthalmic examination based on the International Classification System for Retinoblastoma (Table 12.2). The (International Retinoblastoma Staging System (IRSS) shown in Table 12.3a) is based on histopathology and imaging findings and takes into account that EORB with spread to regional lymphnode (LN) has a rate of overall survival comparable to non metastatic extraocular retinoblastoma (EORB).
Group | Clinical features |
|---|---|
A Very low risk | All tumors are 3 mm or smaller, confined to the retina, and located at least 3 mm from the foveola and 1.5 mm from the optic nerve. |
B Low risk | Retinal tumors may be of any size or location not in Group A. No vitreous or subretinal seeding allowed. A small cuff of subretinal fluid extending no more than 5 mm from the base of the tumor is allowed. |
C Moderate risk | Eyes with only focal vitreous or subretinal seeding and discrete retinal tumors of any size and location. Vitreous or subretinal seeding may extend no more than 3 mm from tumor. Up to 1 quadrant of subretinal fluid may be present. |
D High risk | Eyes with diffuse vitreous or subretinal seeding and/or massive, nondiscrete endophytic or exophytic disease. |
E Very high risk eyes | Eyes with one or more of the following: Neovascular glaucoma Massive intraocular hemorrhage Aseptic orbital cellulites Phthisis or pre-phthisis Tumor anterior to anterior vitreous face Tumor touching the lens Diffuse infiltrating retinoblastoma |
Table 12.3a
International Retinoblastoma Staging System (IRSS)
Stage | Description | |
|---|---|---|
Stage 0 | Eye has not been enucleated and no dissemination of disease | |
Conservative treatment | ||
Stage I | Eye enucleated, completely resected histologically | |
Stage II | Eye enucleated, microscopic residual tumor in form of: | |
(i) Tumor invasion into extrascleral space | ||
(ii) Tumor invasion into cut end of ON | ||
Stage III | Regional extension | (a) Overt orbital disease |
(b) Preauricular or cervical lymph node extension | ||
Stage IV | Metastatic disease | (a) Hematogenous metastasis (without CNS involvement) |
1. Single lesion | ||
2. Multiple lesions | ||
(b) CNS extension (with or without any other site of regional or metastatic disease) | ||
1. Prechiasmatic lesion | ||
2. CNS mass | ||
3. Leptomeningeal and CSF disease | ||
12.1.2.6 Treatment for Retinoblastoma
Principles
The first aim of treatment is survival of the child with preservation of the globe and vision being secondary goals (Table 12.3b). With improved survivorship, focus on cosmesis with the use of prosthetic eye has also become important. Treatment depends on size and location of the tumor and whether it is unilateral or bilateral. Therapeutic plans usually require a multidisciplinary approach. Treatment should be highly individualized. Systemic chemotherapy has become the cornerstone of therapy for retinoblastoma and primarily includes vincristine, carboplatin, and etoposide. Chemotherapy may be administered through local routes also. Systemic chemotherapy may be used for primary chemoreduction, as an adjunct modality or for treatment of metastasis. The major concern is to avoid enucleation and/or external beam radiation, and trends are toward conservative treatment in intraocular disease.
Table 12.3b
Broad principles of chemotherapy
Modality | Schedule | Indication/classification |
|---|---|---|
Chemoreduction for large I/O tumors | 2–6 cycles before consolidative therapy up to 12 cycles | ICRB groups B–D Globe salvation |
Chemoprophylaxis | 6 cycles | Any high-risk histopathological features Invasion of the anterior chamber, iris, ciliary body Massive choroid invasion Invasion of sclera Post-laminar ON invasion |
Adjuvant chemotherapy for microscopic residual disease | (12 cycles) | Extrascleral involvement Invasion of ON reaching cut end |
Neoadjuvant chemotherapy (NACT) | 2–3 cycles | Stage III EORB (orbital mass/ON involvement on MRI OR anterior fungating mass clinically) |
High-dose chemotherapy with autologous SCT | Stage IV/metastatic RB |
In cases of unilateral disease with large tumors where no useful vision can be preserved, enucleation must be performed early; a delay leads to progressive disease. In children with bilateral disease, systemic chemotherapy is used to shrink the tumors, followed by local treatment with transpupillary thermotherapy or cryotherapy in order to preserve vision. Newer routes of drug administration including periocular, intravitreal, and selective intra-arterial chemotherapy have improved outcomes particularly in intraocular retinoblastoma (Shields et al. 1993; Banavali 2004).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


