Cell Structure and Function
Jacquelyn L. Banasik
Key Questions
• What are the major cellular structures and their functions?
• How do cells acquire and use energy?
• How are substances transported across the cell membrane?
• Why is it that some cells can produce action potentials and others cannot?
• How do cells in a multicellular organism communicate with one another?
• What are the normal mechanisms of cellular growth control?

http://evolve.elsevier.com/Copstead/
A basic principle of biology states that the cell is the fundamental unit of life. As more diseases are understood on the cellular and molecular levels, it appears that the cell is also the fundamental unit of disease. A knowledge explosion is currently occurring in the fields of cell and molecular biology, leading to a better understanding of human physiology and the cellular aspects of disease. Detailed knowledge of cellular dysfunction has led to the development of more specific and appropriate prevention and treatment modalities for many disease processes. Thus, an understanding of cellular mechanisms is essential for health care providers and fundamental to the discussions of pathophysiologic processes presented throughout the remainder of this text.
Cells are complex, membrane-bound units packed with a multitude of chemicals and macromolecules. They are able to replicate and thus form new cells and organisms. The very first cells on Earth probably arose from the spontaneous association of organic (carbon-containing) and inorganic molecules about 3.5 billion years ago.1 Over billions of years, the self-replicating molecules now known as deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) are believed to have evolved by chance association and natural selection. Development of the cell membrane created a closed compartment that provided a selective advantage for the cell and accomplished the first separation of life (inside) from nonlife (outside). In this protected environment, the early cells continued to evolve and develop. Today, a large number of different cell types exist, but many of the basic biochemical mechanisms of these cells are remarkably similar. Scientists believe that all modern cells, from bacteria to human neurons, evolved from common primordial cells.2 It is therefore possible to unlock many of the secrets of human cellular physiology by studying easily grown and rapidly proliferating cells, such as yeasts and bacteria.
Much of our knowledge of cell physiology has derived from study of the class of cells known as prokaryotic, which includes bacteria and archaea. Prokaryotic cells are smaller and simpler than eukaryotic cells, having no defined nucleus or cytoplasmic organelles. Fungi, plants, and animals belong to the eukaryotic class of cells, which possess a membrane-bound nucleus and a host of cytoplasmic organelles (Figure 3-1). In this chapter, the essentials of eukaryotic cell structure, physiology, metabolism, and communication are reviewed.
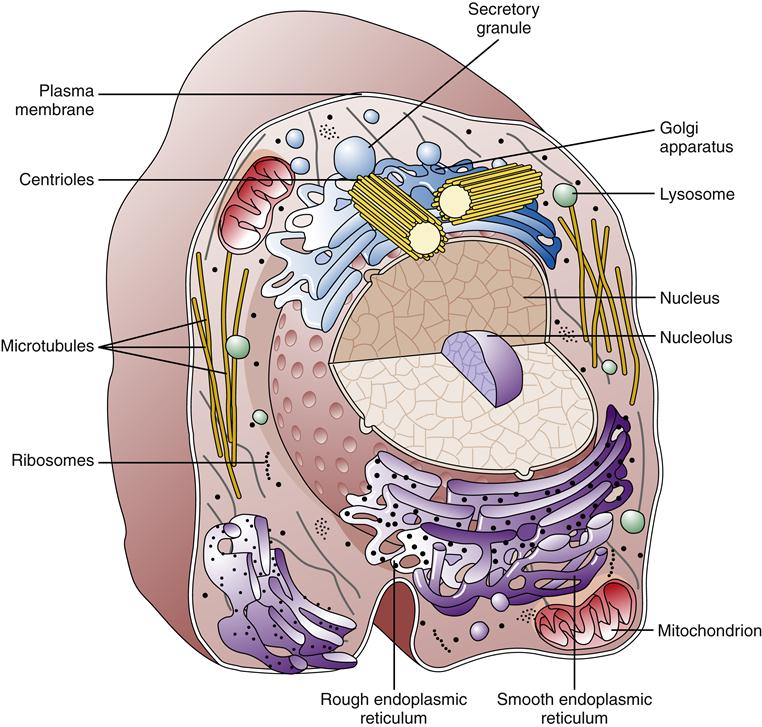
Plasma Membrane
Membrane Structure
All cells are enclosed by a barrier composed primarily of lipid and protein called the plasma membrane (plasmalemma). This cell membrane is a highly selective filter that shields internal cell contents from the external environment. The plasma membrane performs a variety of functions, including transport of nutrients and waste products, generation of membrane potentials, and recognition, communication, and growth regulation of cells. The cell membrane is a sensor of signals and enables the cell to respond and adapt to changes in its environment.
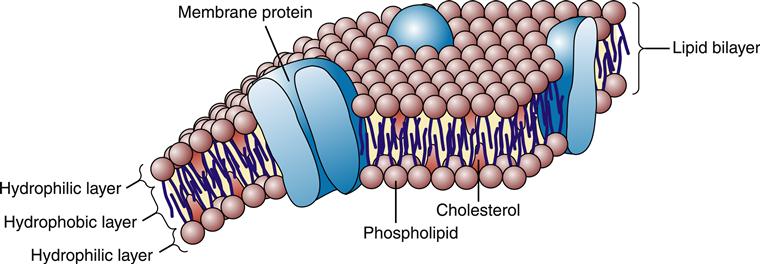
According to the fluid mosaic model first described in the 1960s by Singer and Nicolson,3 the plasma membrane is a dynamic assembly of lipid and protein molecules. Most of the lipids and proteins move about rapidly in the fluid structure of the membrane. As shown in Figure 3-2, the lipid molecules are arranged in a double layer, or lipid bilayer, which is highly impermeable to most water-soluble molecules, including ions, glucose, and proteins. A variety of proteins embedded or “dissolved” in the lipid bilayer perform most of the membrane’s functions. Some membrane proteins are involved in the transport of specific molecules into and out of the cell; others function as enzymes or respond to external signals; and some serve as structural links that connect the plasma membrane to adjacent cells. The lipid structure of the plasma membrane is similar to the structure of the membrane that surrounds the cell’s organelles (e.g., nucleus, mitochondria, endoplasmic reticulum, Golgi apparatus, lysosomes).
Lipid Bilayer
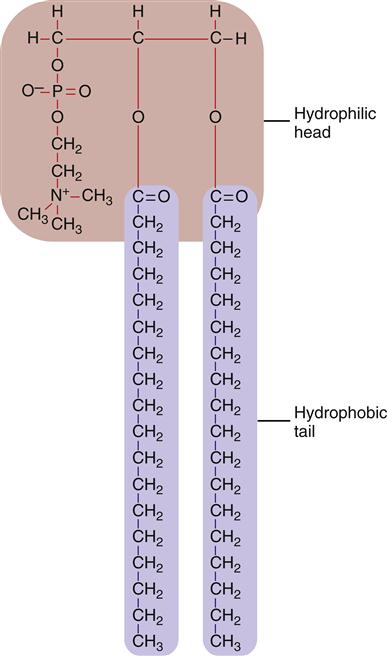
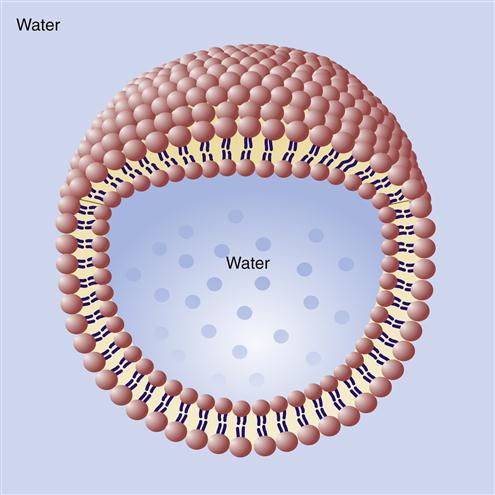
The bilayer structure of all biological membranes is related to the special properties of lipid molecules that cause them to spontaneously assemble into bilayers. The three major types of membrane lipids are cholesterol, phospholipids, and glycolipids. All three have a molecular structure that is amphipathic; that is, they have a hydrophilic (water-loving) charged or polar end and a hydrophobic (water-fearing) nonpolar end.1 This amphipathic nature causes the lipids to form bilayers in aqueous solution. A typical phospholipid molecule is shown in Figure 3-3. The hydrophobic nonpolar tails tend to associate with other hydrophobic nonpolar tail groups to avoid association with polar water molecules. The hydrophilic polar head groups preferentially interact with the surrounding aqueous environment. A bilayer, with tails sandwiched in the middle, allows both portions of the lipid molecules to be chemically “satisfied.” In addition, the lipid bilayers tend to close on themselves, forming sealed, spherical compartments (Figure 3-4). If the membrane is punctured or torn, it will spontaneously reseal itself to eliminate contact of the hydrophobic tails with water.
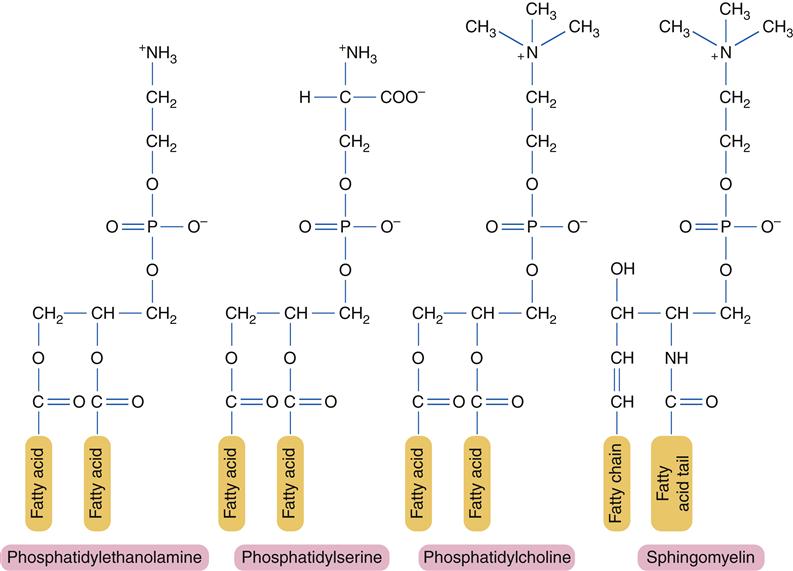
For the most part, individual lipid and protein molecules can diffuse freely and rapidly within the plane of the bilayer. The degree of membrane fluidity depends on the lipid composition. Saturated lipids have straight tails that can pack together and tend to stiffen the membrane, whereas lipids with bent, unsaturated hydrocarbon tails tend to increase fluidity. About 50% of the lipid in eukaryotic cell membranes is cholesterol, which serves to decrease membrane permeability and prevent leakage of small water-soluble molecules. In addition to affecting fluidity by the degree of saturation of tail groups, the phospholipids that inhabit the membrane also differ in the size, shape, and charge of the polar head groups. Figure 3-5 shows the structures of the four most prevalent membrane phospholipids: phosphatidylethanolamine, phosphatidylserine, phosphatidylcholine, and sphingomyelin. Some membrane-bound proteins require specific phospholipid head groups to function properly. Some lipids—sphingolipids and cholesterol in particular—may bind together transiently to form rafts in the sea of moving lipids. These rafts may surround and help organize membrane proteins into functional units. For example, a membrane receptor and its intracellular target proteins may associate together in a raft to facilitate transfer of information across the membrane.4
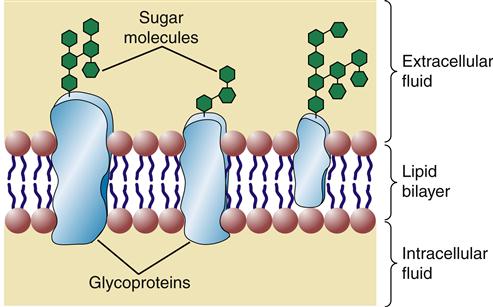
Glycolipids contain one or more sugar (i.e., carbohydrate) molecules at the polar head region. Glycolipids and glycoproteins are found only in the outer half of the lipid bilayer, with the sugar groups exposed at the cell surface (Figure 3-6). Membrane glycolipids are involved in cell recognition and cell-to-cell interactions.5
Membrane Proteins
Approximately 50% of the mass of a typical cell membrane is composed of protein. The specific types of membrane proteins vary according to cell type and environmental conditions. Some membrane proteins, called transmembrane proteins, extend across the membrane bilayer and are in contact with both the extracellular and the intracellular fluids. Transmembrane proteins serve a variety of functions, including transport of charged and polar molecules into and out of cells and transduction of extracellular signals into intracellular messages. Other peripheral membrane proteins are less tightly anchored to the membrane. The common structural orientations of membrane proteins are shown in Figure 3-7. The amino acid structure of membrane proteins determines the way they are arranged in the membrane. Nonpolar amino acids tend to inhabit the hydrophobic middle of the membrane, whereas charged and polar amino acids protrude into the aqueous fluid or associate with polar lipid head groups. The three-dimensional structure of many membrane proteins is complex, with numerous twists and turns through the lipid bilayer.
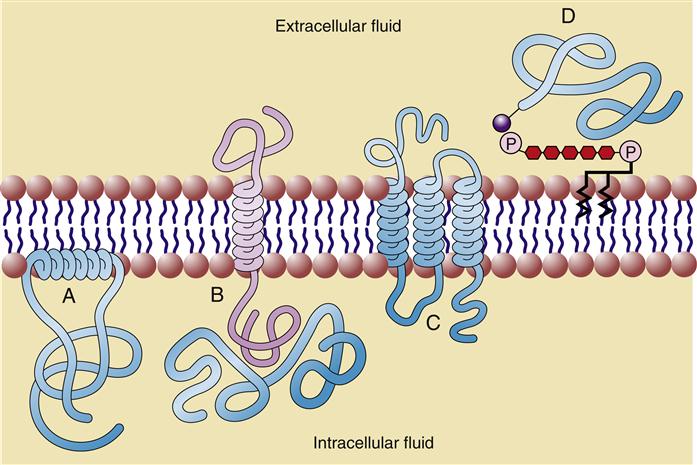
The type of membrane proteins in a particular cell depends on the cell’s primary functions. For example, a kidney tubule cell has a large proportion of transmembrane proteins, which are needed to perform the kidney’s function of electrolyte and nutrient reabsorption. In contrast, the human red blood cell (RBC) contains mainly peripheral proteins attached to the inner surface of the membrane.6 One of these proteins, spectrin, has a long, thin, flexible rodlike shape that forms a supportive meshwork or cytoskeleton for the cell. It is this cytoskeleton that enables the RBC to withstand the membrane stress of being forced through small capillaries.
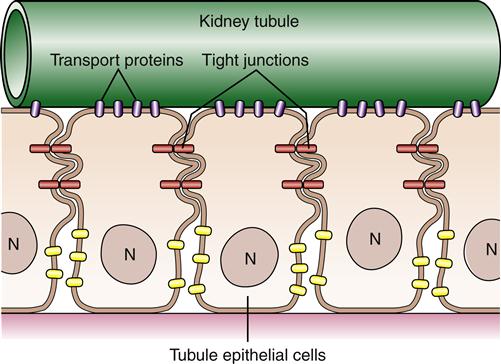
Although proteins and lipids are generally free to move within the plane of the cell membrane, many cells are able to confine certain proteins to specific areas. Using the example of the kidney tubule cell again, it is important for the cell to keep transport proteins on its luminal side to reabsorb filtered molecules (Figure 3-8). This segregation of particular proteins is accomplished primarily by intercellular connections called tight junctions, which connect neighboring cells and function like a fence to confine proteins to an area of the membrane. Membrane proteins also can be immobilized by tethering them to cytoskeleton or extracellular matrix structures.
Organization of Cellular Compartments
Cytoskeleton
Eukaryotic cells have a variety of internal compartments, or organelles, that are membrane bound and carry out distinct cellular functions. The cell’s organelles are not free to float around haphazardly in the cytoplasmic soup; rather, they are elaborately organized by a protein network called the cytoskeleton (Figure 3-9).7 The cytoskeleton maintains the cell’s shape, allows cell movement, and directs the trafficking of substances within the cell. Three principal types of protein filaments make up the cytoskeleton: actin filaments, microtubules, and intermediate filaments.
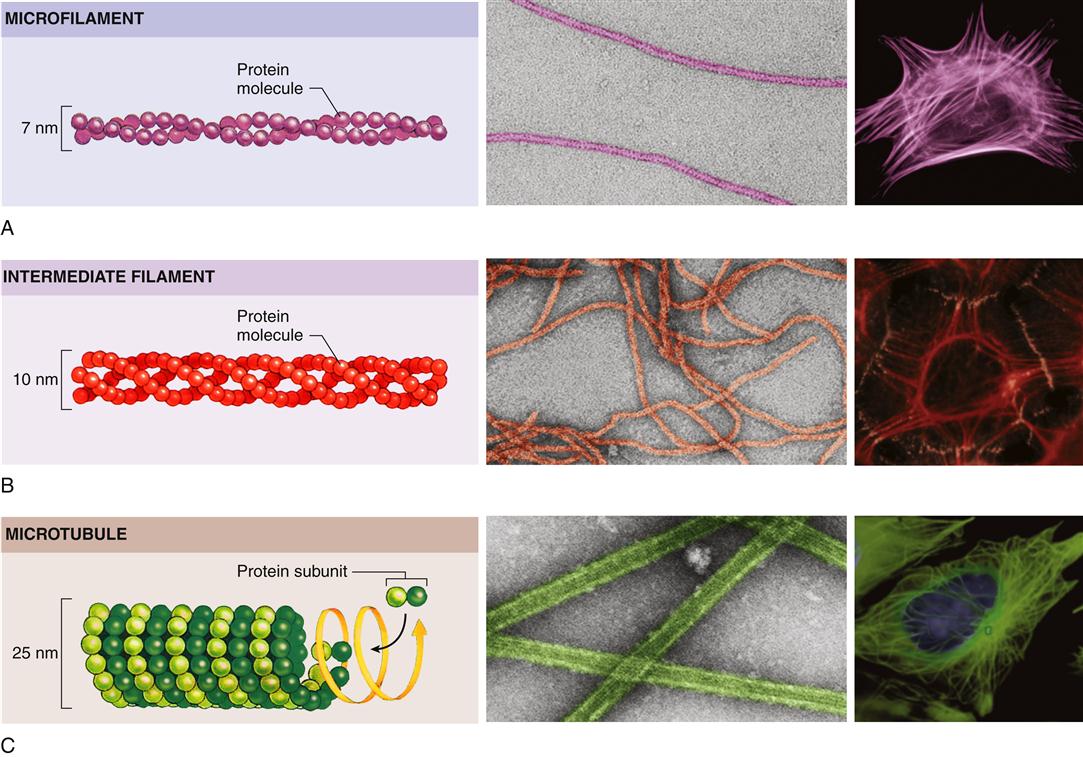
All three types of filaments consist of small proteins that can assemble (polymerize) into filaments of varying length. The filament structures are dynamic and can be rapidly disassembled and reassembled according to the changing needs of the cell. Actin filaments play a pivotal role in cell movement. As one might expect, muscle cells are packed with actin filaments, which allows the cell to perform its primary function of contraction. However, nonmuscle cells also possess actin filaments that are important for complex movements of the cell membrane, such as cell crawling and phagocytosis. Such movements of the cell membrane are mediated by dense networks of actin filaments that cluster just beneath the plasma membrane and interact with specific proteins embedded in it. Actin and some of the other cytoskeletal proteins make specific contacts with and through the plasma membrane and are involved in information transfer from the extracellular environment to signaling cascades within the cell.
Organization of the cytoplasm and its organelles is achieved primarily by microtubules. In animal cells, microtubules originate at the cell center, or centrosome, near the nucleus and radiate out toward the cell perimeter in fine lacelike threads. Microtubules guide the orderly transport of organelles and vesicles in the cytoplasm as well as the equal distribution of chromosomes during cell division. Intermediate filaments, so named because their size is between that of microtubules and actin filaments, are strong, ropelike, fibrous proteins. A variety of intermediate filaments that differ from tissue to tissue have been identified. In addition to the three main groups of cytoskeletal filaments just described, a large number of accessory proteins are essential for cytoskeletal function. For example, the accessory protein myosin is needed to bind with actin to achieve motor functions. Different accessory proteins are present in different cell types.
Nucleus
The largest cytoplasmic organelle is the nucleus, which contains the genetic information for the cell in the form of DNA. The human genome contains approximately 23,000 genes that code for proteins, representing less than 1.3% of the total DNA structure composed of more than 6 billion base pairs.8 The nuclear contents are enclosed and protected by the nuclear envelope, which consists of two concentric membranes. The inner membrane forms an unbroken sphere around the DNA and contains protein-binding sites that help to organize the chromosomes inside. The outer nuclear membrane is continuous with the endoplasmic reticulum (ER) (see next section) and closely resembles it in structure and function (Figure 3-10). The nucleus contains many proteins that help mediate its functions of genetic control and inheritance. These proteins, including histones, polymerases, and regulatory proteins, are manufactured in the cytosol and transported to the nucleus through holes in the membrane called nuclear pores. The nuclear pores are selective as to which molecules are allowed access to the nuclear compartment, and in this way they protect the genetic material from enzymes and other molecules in the cytoplasm. The nuclear pores also mediate the export of products such as RNA and ribosomes that are synthesized in the nucleus but function in the cytosol. Ribosomes are manufactured in a specialized portion of the nucleus called the nucleolus. Nuclear pores are complexes of proteins that span across both the inner and the outer nuclear membrane, creating a pathway between the cytoplasm and the nuclear lamina (see Figure 3-10).
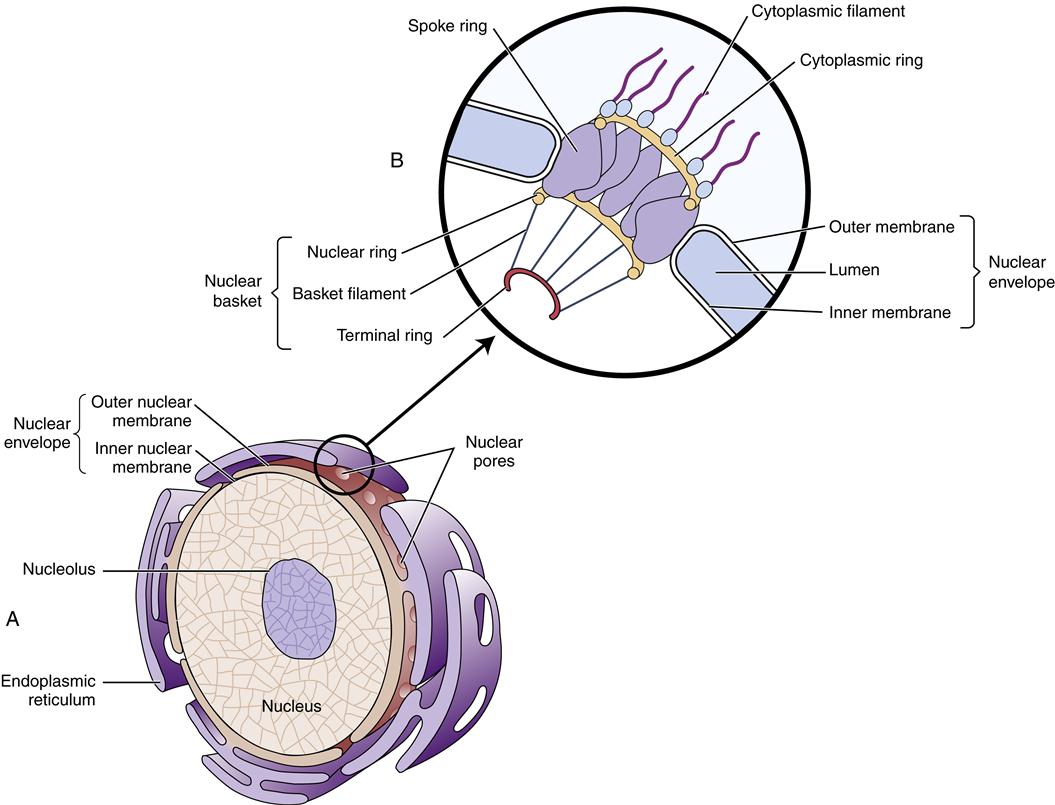
A major function of the nucleus is to protect and preserve genetic information so that it can be replicated exactly and passed on during cell division. However, the nucleus is continuously functioning even when the cell is not actively dividing. The nuclear DNA controls the production of cellular enzymes, membrane receptors, structural proteins, and other proteins that define the cell’s type and behavior. (The structure and function of DNA are discussed in Chapter 5.)
During mitosis, the complex structure of the nuclear membrane and its pore-forming proteins breaks into small pieces that diffuse through the cell cytoplasm. After cell division is complete, pieces of nuclear membrane surround and gather the chromosomes and then fuse together to form a new nuclear membrane. Nuclear proteins and pore structures are then recruited back to their normal nuclear locations.9
Endoplasmic Reticulum
The ER is a membrane network that extends throughout the cytoplasm and is present in all eukaryotic cells (Figure 3-11). The ER is thought to have a single continuous membrane that separates the lumen of the ER from the cytosol—it could be likened to a “gastrointestinal tract” in the cell. The ER plays a central role in the synthesis of membrane components, including proteins and lipids, for the plasma membrane and cellular organelles, as well as in the synthesis of products to be secreted from the cell. The ER is divided into rough and smooth types based on its appearance under the electron microscope. The rough ER is coated with ribosomes along its outer surface. Ribosomes are complexes of protein and RNA that are formed in the nucleus and transported to the cytoplasm. Their primary function is the synthesis of proteins (see Chapter 5). Depending on the destination of the protein to be created, ribosomes may float free in the cytosol or may bind to the ER membrane. Proteins synthesized by free-floating ribosomes are released within the cytosol of the cell. Proteins to be transported into the ER have a special sequence of amino acids that directs the ribosome responsible for its synthesis to the ER membrane. Special proteins called signal recognition particles (SRPs) bind to the leading sequence of the protein and then bind to a receptor on the ER membrane. As the ribosome adds amino acids to the growing protein chain, it is pushed into the lumen of the ER through a pore in the ER membrane called a translocon.10 After being processed in the ER and Golgi apparatus, the protein is eventually transported to the appropriate organelle or secreted at the cell surface. Free-floating and rough ER ribosomes are identical and interchangeable; their location depends on the amino acid structure of the protein they are producing at the time.11
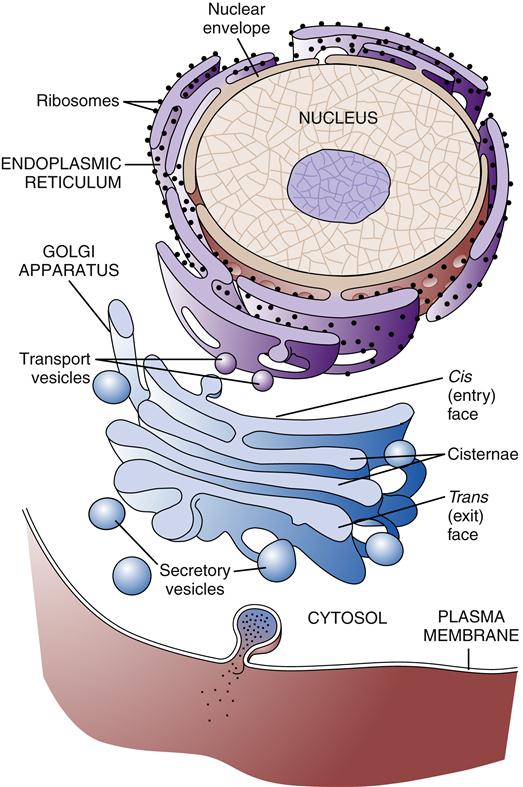
Regions of ER that lack ribosomes are called smooth ER. The smooth ER is involved in lipid metabolism. Most cells have very little smooth ER, but cells specializing in the production of steroid hormones or lipoproteins may have significant amounts of smooth ER. For example, the hepatocyte (liver cell) has abundant smooth ER–containing enzymes (P450) responsible for the manufacture of lipoproteins as well as the detoxification of harmful lipid-soluble compounds, such as alcohol. The cellular smooth ER can double in surface area within a few days if large quantities of drugs or toxins enter the circulation. Cells in the adrenal cortex and gonads that produce steroid hormones also have abundant smooth ER. In addition to synthetic functions, the ER also sequesters large amounts of calcium ions by pumping them from the cytoplasm. In response to specific signals, the ER releases calcium ions as part of important second-messenger cascades. Muscle cells have extensive smooth ER (sarcoplasmic reticulum) dedicated to the sequestration of calcium. When the cell is stimulated, the sarcoplasmic reticulum releases the calcium ions needed to accomplish muscle contraction.
Golgi Apparatus
The Golgi apparatus or Golgi complex is composed of a stack of smooth membrane-bound compartments resembling a stack of hollow plates (see Figure 3-11). These compartments or cisternae are organized in a series of at least three processing compartments. The first compartment (cis face) lies next to the ER and receives newly synthesized proteins and lipids by way of ER transport vesicles. These transport vesicles are outgrowths that bud off from the ER membrane and diffuse to the Golgi, where they bind and become part of the Golgi apparatus membrane. The proteins and lipids then move through the middle compartment (medial) to the final compartment (trans face), where they depart for their final destination. As the lipid and protein molecules pass through the sequence of Golgi compartments, they are modified by enzymes that attach or rearrange carbohydrate molecules. After specific arrangement of these carbohydrates has occurred, the lipids and proteins are packaged into Golgi transport vesicles (secretory vesicles). The particular configuration of carbohydrate molecules on the lipid or protein is believed to serve as an “address label,” directing them to the correct destination within the cell. Golgi vesicles transport their contents primarily to the plasma membrane and to lysosomes.
Lysosomes and Peroxisomes
Transport of Golgi vesicles to the membrane-bound bags of digestive enzymes known as lysosomes has been well described and provides a model for Golgi sorting and transport to other destinations. Lysosomes are filled with more than 40 different acid hydrolases, which are capable of digesting organic molecules, including proteins, nucleotides, fats, and carbohydrates.12 Lysosomes obtain the materials they digest from three main pathways. The first is the pathway used to digest products absorbed by endocytosis. In this pathway, endocytotic vesicles bud off from the plasma membrane to fuse with endosomes. Endosomes mature into lysosomes as the Golgi delivers lysosomal enzymes to them; the pH inside the lysosome acidifies, and active digestion occurs. The second pathway is autophagy, whereby damaged and obsolete parts of the cell itself are destroyed. Unwanted cellular structures are enclosed by a membrane from the ER, which then fuses with the lysosome, leading to autodigestion of the cellular components. Autophagy also may occur during cell starvation or disuse, leading to a process called atrophy, in which the cell becomes smaller and more energy efficient. The third pathway providing materials to the lysosomes is present only in specialized phagocytic cells. White blood cells (WBCs), for example, are capable of ingesting large particles, which then form a phagosome capable of fusing with a lysosome. The final products of lysosomal digestion are simple molecules, such as amino acids, fatty acids, and carbohydrates, which can be used by the cell or secreted as cellular waste at the cell surface.
Discovery of the mechanism for sorting and transport of lysosomal enzymes was aided by studying patients suffering from the lysosomal storage diseases.13 Patients with I-cell (inclusion cell) disease, for example, accumulate large amounts of debris in lysosomes, which appear as spots or “inclusions” in the cells. These lysosomes lack nearly all of the hydrolases normally present and thus are unable to perform lysosomal digestion. However, all the hydrolases missing from the lysosomes can be found in the patient’s bloodstream. The abnormality results from “mis-sorting” by the Golgi apparatus, which erroneously packages the enzymes for extracellular secretion rather than sending them to the lysosomes. Studies of this rare genetic disease resulted in the discovery that all lysosomal enzymes have a common marker, mannose-6-phosphate, which normally targets the enzymes to the lysosomes. Persons with I-cell disease lack the enzyme responsible for attaching this marker.
Peroxisomes (microbodies), like lysosomes, are membrane-bound bags of enzymes that perform degradative functions. They are particularly important in liver and kidney cells, where they detoxify various substances, such as alcohol. In contrast to lysosomes, which contain hydrolase enzymes, peroxisomes contain oxidative enzymes. These enzymes use molecular oxygen to break down organic substances by an oxidative reaction that produces hydrogen peroxide. The hydrogen peroxide is then used by another enzyme (catalase) to degrade other organic molecules, including formaldehyde and alcohol. Catalase also prevents accumulation of excess hydrogen peroxide in the cell by converting it to water and oxygen. Peroxisomes also oxidize fatty acids (β oxidation) to produce acetyl coenzyme A (acetyl CoA) that is used in cellular metabolism. Unlike lysosomes, which acquire their enzymes from Golgi vesicles, peroxisomes import enzymes directly from the cytoplasm.
Mitochondria
The mitochondria have been aptly called the “powerhouses of the cell” because they convert energy to forms that can be used to drive cellular reactions. A distinct feature of mitochondria is the large amount of membrane they contain. Each mitochondrion is bound by two specialized membranes. The inner membrane forms an enclosed space, called the matrix, which contains a concentrated mix of mitochondrial enzymes. The highly convoluted structure of the inner membrane with its numerous folds, called cristae (Figure 3-12), provides a large surface area for the important membrane-bound enzymes of the respiratory chain. These enzymes are essential to the process of oxidative phosphorylation, which generates most of the cell’s adenosine triphosphate (ATP). The outer membrane contains numerous porin transport proteins forming large aqueous channels that make the membrane porous like a sieve. Fairly large molecules, including proteins up to 5000 daltons, can pass freely through the outer membrane such that the space between the outer and inner membranes is chemically similar to the cytosol. However, the inner membrane is quite impermeable, even to small molecules and ions. Specific protein transporters are required to shuttle the necessary molecules across the inner mitochondrial membrane.
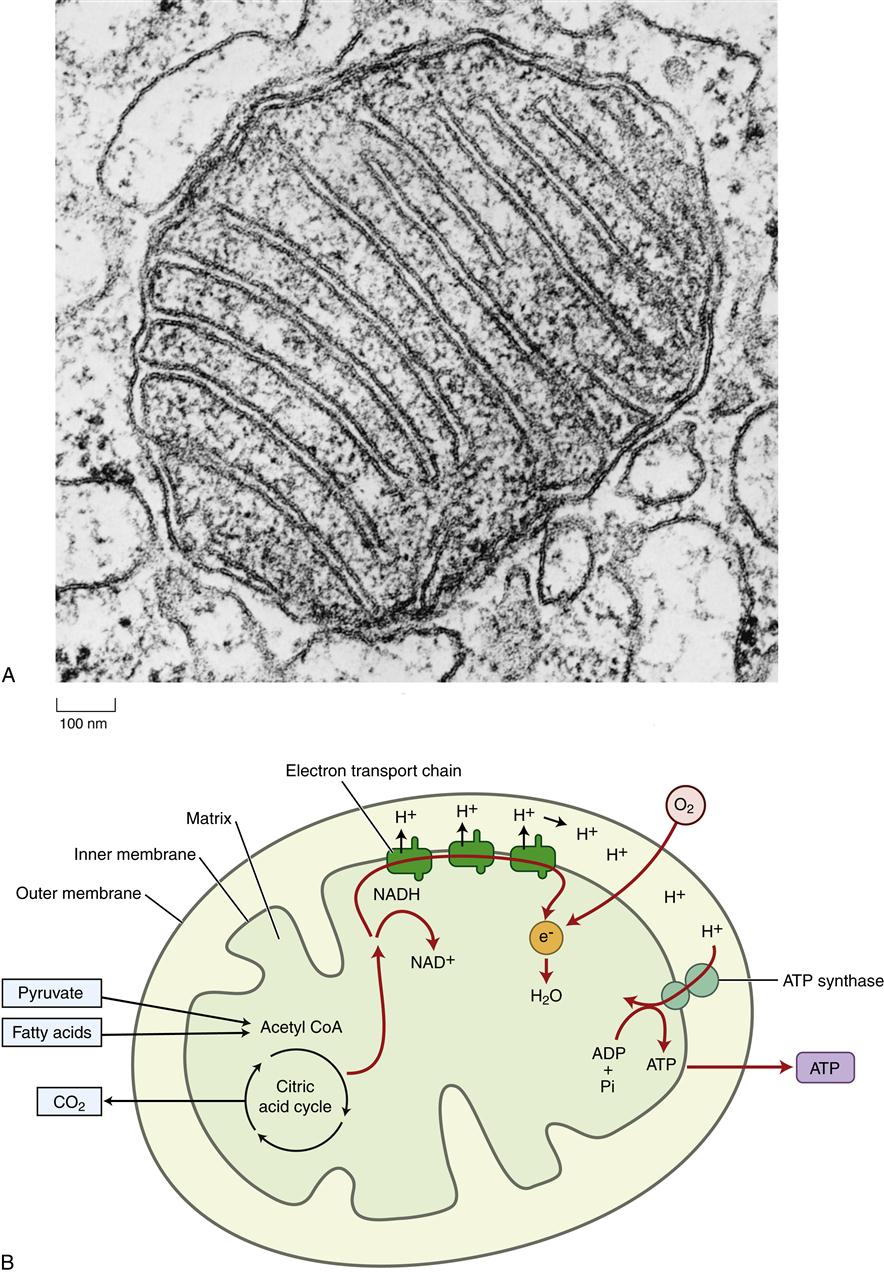
Mitochondria are believed to have originated as bacteria that were engulfed by larger cells but that still retain some of their own DNA. Mitochondrial DNA codes for 22 transfer RNA molecules, 2 ribosomal RNAs that form mitochondrial ribosomes, and 13 proteins.14–16 During evolution the majority of mitochondrial genes were transferred to locations within the nuclear genome. Thus only a few of the mitochondrial enzymes are produced from DNA located in the mitochondria; the majority are transcribed from nuclear DNA. Nuclear genes are translated into protein in the cytoplasm and then transported to the mitochondria, whereas mitochondrial gene–derived proteins are made within the mitochondria. There are several rare disorders associated with mitochondrial gene defects (see Chapter 6). The number and location of mitochondria differ according to cell type and function. Cells with high energy needs, such as cardiac or skeletal muscle, have many mitochondria. These mitochondria may pack between adjacent muscle fibrils, such that ATP is delivered directly to the areas of unusually high energy consumption. The details of mitochondrial energy conversion are discussed in the next section. Mitochondria also have an important role in programmed cell death, called apoptosis, which is discussed in Chapter 4.
Cellular Metabolism
All living cells must continually perform essential cellular functions such as movement, ion transport, and synthesis of macromolecules. Many of these cellular activities are energetically unfavorable (i.e., they are unlikely to occur spontaneously). Unfavorable reactions can be driven by linking them to an energy source such as ATP, which is a molecule that contains high-energy phosphate bonds. In normal cells where the ATP concentration is high, approximately 11 to 13 kcal of energy per mole of ATP is liberated when one of the phosphate bonds is hydrolyzed (broken with the aid of water) in a chemical reaction.15 Enzymes throughout the cell are able to capture the energy released from ATP hydrolysis and use it to break or make other chemical bonds. In this way, ATP serves as the “energy currency” of the cell. A specific amount of ATP is “spent” to “buy” a specific amount of work. Most cells contain only a small amount of ATP, sufficient to maintain cellular activities for just a few minutes. Because ATP cannot cross the plasma membrane, each cell must continuously synthesize its own ATP to meet its energy needs; ATP cannot be “borrowed” from other cells or “banked” in any significant quantity within a cell. ATP is synthesized primarily from the breakdown of glycogen and fat.
An average adult has enough glycogen stores (primarily in liver and muscle) to supply about 1 day’s needs, but enough fat to last for a month or more. After a meal, the excess glucose entering the cells is used to replenish glycogen stores or to synthesize fats for later use. Fat is stored primarily in adipose tissue and is released into the bloodstream for other cells to use when needed. When cellular glucose levels fall, glycogen and fats are broken down to provide glucose and fatty acyl molecules, respectively, which are ultimately metabolized to provide ATP. During starvation, body proteins can also be used for energy production by a process called gluconeogenesis.
Cellular metabolism is the biochemical process whereby foodstuffs are used to provide cellular energy and biomolecules. Cellular metabolism includes two separate and opposite phases: anabolism and catabolism. Anabolism refers to energy-using metabolic processes or pathways that result in the synthesis of complex molecules such as fats. Catabolism refers to the energy-releasing breakdown of nutrient sources such as glucose to provide ATP to the cell. Both of these processes require a long, complex series of enzymatic steps. The catabolic processes of cellular energy production are briefly discussed in the following sections. (See Chapter 42 for a detailed discussion of metabolism.)
Glycolysis
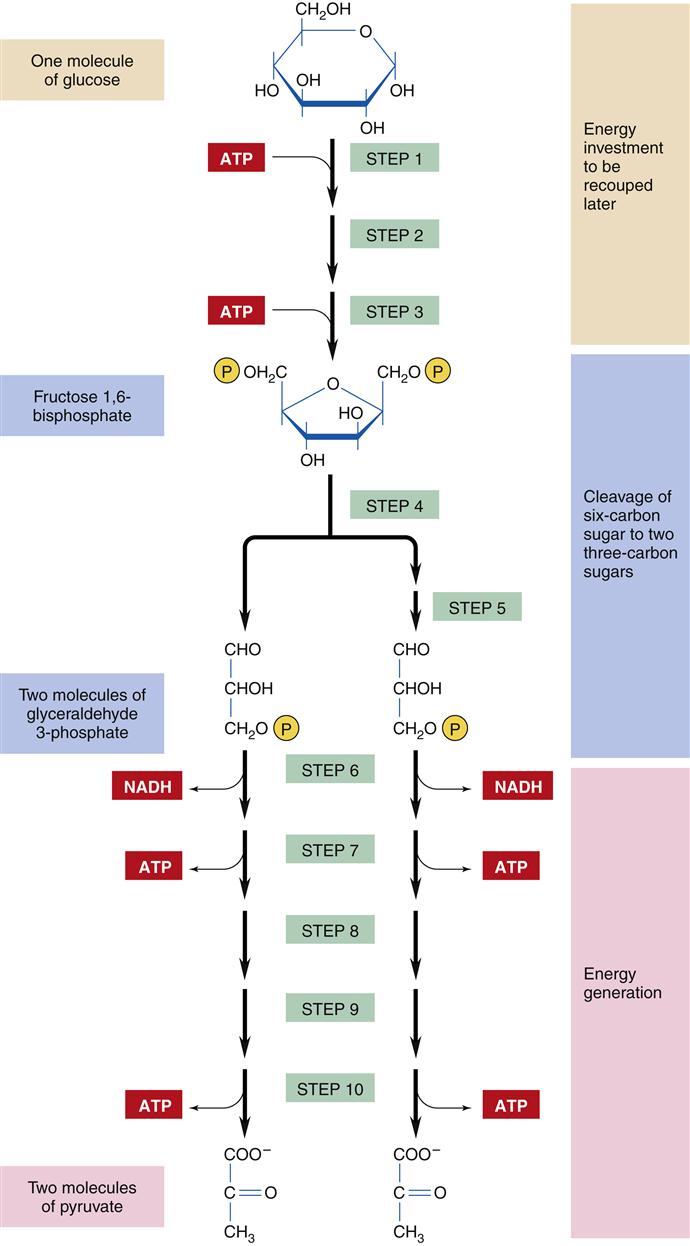
The catabolic process of energy production begins with the intestinal digestion of foodstuffs into small molecules: proteins into amino acids, polysaccharides into simple sugars (monosaccharides), and fats into fatty acids and glycerol. The second stage of catabolism occurs in the cytosol of the cell, where glucose molecules are further degraded by glycolysis into pyruvate (compounds with three carbon atoms). Glycolysis involves 10 enzymatic steps to break the 6-carbon glucose molecule into a pair of 3-carbon pyruvate molecules (Figure 3-13).15 Glycolysis requires the use of two ATP molecules in the early stages but produces four ATP molecules in the later steps, for a net gain of two ATP molecules per glucose molecule. The production of ATP through glycolysis is relatively inefficient, and the pyruvate end products still contain substantial chemical energy that can be released by further catabolism in stage 3. However, glycolysis is an important provider of ATP under anaerobic conditions because oxygen is not required. Thus, ATP production by glycolysis becomes important during conditions of reduced cellular oxygenation, which may accompany respiratory and cardiovascular disorders. The pyruvate that accumulates during prolonged anaerobic conditions is converted to lactate and excreted from the cell into the bloodstream. Lactic acidosis is a dangerous condition that may result from excessive lactate production attributable to severe or prolonged lack of oxygen (see Chapter 20). In addition to the two molecules of ATP and pyruvate, each glucose molecule produces two reduced nicotinamide adenine dinucleotide (NADH) molecules, which contain high-energy electrons that are transferred to the electron transport chain in the mitochondria. Cells that do not contain mitochondria, such as RBCs, must rely totally on glycolysis for ATP production.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


