THE VASCULAR RESPONSE TO INJURY
GENERAL INTRODUCTION
A long tradition defines the scope of pathology as both a clinical specialty and an area of biomedical research. Although rooted in the correlation of anatomical and histological changes with clinically apparent disease (and hence the iconic images of autopsy and microscope), modern pathology studies the causes of disease (etiology) and the expression/evolution of such (pathogenesis) at the molecular level using the tools of molecular genetics and biochemical analysis as well as at the cellular and organ system levels. Because (as is apparent to the clinician) the first symptoms and signs of disease are often those of the body’s response to injury, the pathologist is acutely interested in characterizing this response since, more often than not, such provides critical clues as to the etiological agent and likely mode of pathogenesis.
As a clinical endeavor, pathology is both a diagnostic and prognostic specialty, which by defining and classifying the disease process, hopes to suggest (and help evaluate) therapeutic approaches to the physician. But in a broad sense, pathology seeks to understand the basis of the disease process. At times, this broad scope of interest leads to a “pathocentric” view of medicine (Figure 2-1).
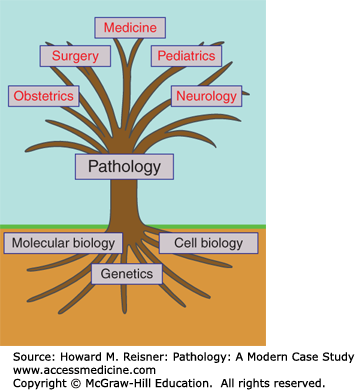
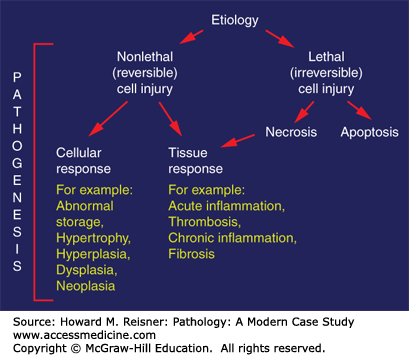
This chapter will present an overview of how the pathologist views mechanisms of irreversible cell injury (cell death), reversible cell injury, and the organism’s response to both. Oxygen deprivation to tissue (ischemia) leading to a form of tissue damage termed an infarct is of notable clinical significance (e.g., in myocardial infarcts, “heart attacks”) and will serve as an important model. Second, this chapter will briefly consider injury resulting from the process of host defense, either as appropriately targeted to tissue injury or inappropriately directed toward self-components either as “bystander effects” or “autoimmunity.” Finally, the chapter will consider changes due to the aging process. A simplistic, but nevertheless useful overview of this chapter is presented in Figure 2-2.
CELL INJURY AND CELL DEATH: AN INTRODUCTION
An interest in the effects of disease and trauma is nothing new. People have been pathologists observing the effects of disease since the dawn of recorded history, likely before. Egyptian medical texts described infectious diseases (tetanus is an often quoted example). The Tanach (Hebrew Bible) tells of a disease visited upon the Philistines that may well have been bubonic plague. The Iliad describes the surgeon Machaon treating Menaleus’ arrow wound. Egyptian physicians described the host reaction to such wounds (suppuration, inflammation) as early as the third millennium BC and may have treated such wounds with antiseptic agents. Pathologists now study the cause (etiology), progression (pathogenesis), and host reaction to disease as a career in order to diagnose the illness, prognosticate, help suggest the best therapy, and ultimately discover the cause of death.
Death at the level of the organism (somatic death) would seem easy to define but it is not. This uncertainty has been historically expressed as fear of premature burial, a likely source of the custom of wakes and attendance on the recently dead. In 18th century, France brass pins under finger and toenails was a test for death, but the odor of decay was held more certain (a reasonable sign of postmortem change). Currently, brain death (irreversible coma) is generally accepted as the legal/clinical definition of somatic death. In a multicellular organism, cells die at different rates (depending in part on their particular requirement for oxidative metabolism). With the cessation of heartbeat, cerebral neurons, lacking oxygen, die within minutes, muscle, skin, and bone cells may survive for days. “Cessation of all life at the cellular level is not a necessary criterion for determination of death” (World Medical Association Declaration on Death). Hence, the medical determination of death relies on clinical judgment supplemented by a variety of neurological, physiological, and imaging techniques aimed at defining the cause and irreversibility of coma. Brain-dead individuals are respirator dependent, are areflexive and lack deep intracranial electrical activity. Removed from the respirator blood pressure collapses, oxidative metabolism ceases, and postmortem changes begin. ATP-dependent cellular processes such as the calcium transporting ATPases in muscle sarcomeres fail. The diffusion of calcium inward produces unrelieved muscle contraction termed rigor mortis ceasing only with the onset of decomposition of cells by cellular enzymes and bacteria.
Many societies have tried to prevent or slow postmortem decomposition. Mummification, the drying or embalming of the corpse as practised in pharaonic Egypt for reasons of religion, led to the understanding that the root of decomposition was the gut, which was removed prior to the application of chemical drying agents and preservatives. Observations made during the process of mummification led Egyptian concepts of disease to center around the anus, presumably because of what we now know to be the role of colonic bacterial flora in early decomposition. Based on these observations, an Egyptian pathologist might have considered rot as the etiological agent of disease, pathogenesis as the spread of rot from its source (the anus) through the body.
Contemporary pathologists maintain an interest in somatic death and injury, although as will be discussed in this chapter the focus has shifted to cellular death and injury and the molecular mechanisms associated with such. Nevertheless, the forensic pathologist interested in force injury still must deal with the manner and cause of death in cases of trauma where much or all of the body is affected. From the 14th century forward, the increasing popularity of anatomical dissection in Europe focused interest on abnormalities of organs and organ systems. The process of correlation of visible abnormality with disease continued through the 19th century and continues even today. The perfection of the microscope combined with the development of histological procedures for the fixation, sectioning, and staining of tissue by the late 19th century established our current emphasis on the expression of disease in tissue and cells, an emphasis now combined with genomic and proteomic pathology.
This chapter will discuss the visible signs of cell injury and death and the consequent host response using the well-established tools of the surgical pathologist who examines biopsy material (tissue taken from the living patient) to establish diagnosis or autopsy material to establish or confirm the cause of death. But the chapter will also summarize the molecular mechanisms associated with cell injury, death, and the host response to demonstrate how knowledge of basic mechanisms may translate into a better understanding of etiology and pathogenesis and improved patient care and perhaps a better understanding of the inevitable process of aging.
An all too common clinical scenario will serve to introduce many of the concepts presented in this chapter (see Case 2-1).
REVERSIBLE CELL INJURY
It is convenient to divide the effects of cell injury as being irreversible (lethal) or potentially reversible. However, there is no well-defined “point of no return” separating the two. Although pathologists most commonly use the morphological pattern of tissue injury to divide reversible from lethal injury, biochemical and molecular analysis is increasingly useful. Case 2-1 notes the use of blood-based tests indicative of the death of specific cell populations (cardiac myocytes and hepatocytes being commonly used examples). Evidence for loss of nuclear DNA, either as noted by histological signs of nuclear fragmentation and loss (karyorrhexis and karyolysis) or biochemical analysis indicating DNase-mediated cleavage of DNA, would be obvious evidence of irreversible cell injury.
CASE 2-1
John Doe, a 63-year-old obese man with a long history of cardiovascular disease, was drinking a double mocha frappe with extra whipped cream at his favorite coffee shop when he turned to the barista while clutching his left upper chest. He complained of crushing pain, was obviously diaphoretic (sweaty), and rapidly collapsed. The emergency response team arrived in 10 minutes, at which time Mr. Doe was unconscious and had no detectable pulse or heartbeat. Attempts to restore cardiac function at the scene and subsequently in the emergency room were unsuccessful and Mr. Doe was pronounced dead by the attending emergency room physician.
Given the frequency of occurrence of this particular mechanism of unattended sudden, presumably cardiac death (and Mr. Doe’s prior medical history), it is unlikely that the medical examiner/forensic pathologist would have interest in requesting an autopsy (see Chapter 3). However, because this common scenario demonstrates many important principles discussed in this chapter, let us examine the temporal progression, etiology, and pathogenesis that led to Mr. Doe’s pattern of irreversible cardiac myocyte injury (cellular necrosis) and ultimately somatic death.
John Doe at age 40 was diagnosed as having moderate hypertension. He was advised to lose weight, exercise, and prescribed a regimen of antihypertensive drugs (which he ignored claiming they made him feel tired and dizzy).
At age 50 Mr. Doe complained of intermittent chest pain on exertion that subsided on rest. His physician noted an enlarged left ventricular profile on plain chest film. Mr. Doe submitted to a supervised stress test. Resultant chest pain subsided on administration of a vasodilator (sublingual nitroglycerin). Mr. Doe was diagnosed with left ventricular hypertrophy subsequent to untreated hypertension and stable angina pectoris (chest pain resulting from reversible cardiac muscle ischemia most often related to compromise of blood flow in the coronary arteries). The angina was presumed to result from coronary vascular disease, atherosclerosis. Mr. Doe was placed on an aggressive course of antihypertensive and blood lipid reducing agents and provided with nitroglycerin tabs. He refused all invasive procedures aimed at evaluating the extent of, and possibly treating, his coronary vascular disease. If one were able to biopsy Mr. Doe’s left ventricle, we would likely have observed the following (Figure 2-3).
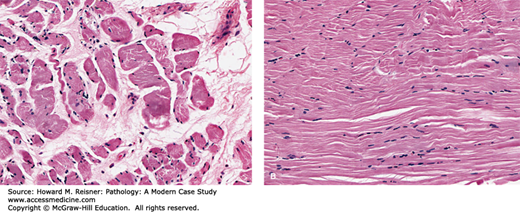
Figure 2-3A demonstrates normal cardiac myocytes; and Figure 2-3B shows hypertrophic cardiac myocytes taken from the heart of an individual with left ventricular hypertrophy. Both views are at the same magnification. In response to physiological stress, cells may either divide and increase in total number (hyperplasia) or, in the case where cell division cannot occur (as in cardiac myocytes), cells may increase in size alone (hypertrophy). In the case of Mr. Doe, the ventricle had to pump against a significantly elevated systemic blood pressure increasing the cardiac workload. Cardiac myocytes compensated by increasing in size. Note that although cardiac myocytes do not divide, they undergo endomitosis in which the average ploidy of the cells increases in the absence of cell division. The resultant increased nuclear size (so-called “boxcar” nuclei) is striking.
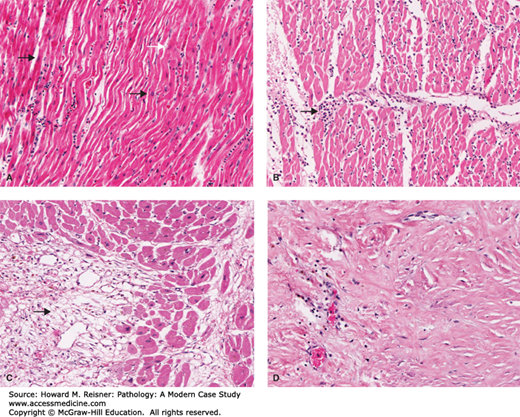
At age 58 Mr. Doe suffered from an episode of crushing chest pain that was unrelieved by nitroglycerine. He was taken to the hospital emergency room where blood tests for cardiac biomarkers troponin I and CK-MB indicated elevated levels consistent with cardiac myocyte cell death (necrosis). Such markers are proteins with specificity for cardiac muscle cells whose release into the systemic circulation is indicative loss of myocyte cell integrity. A severe myocardial infarct was diagnosed and treated. Mr. Doe survived his cardiac injury. If a series of biopsies were available during the time course of the infarct, they would have shown the following (Figure 2-4).
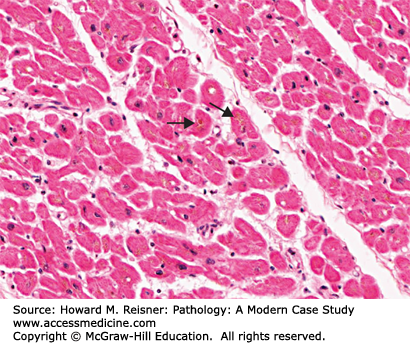
Figure 2-4A (about 12 hours) shows coagulative necrosis of cardiac myocytes. Some cells are eosinophilic and show evidence of nuclear changes ranging from partial dissolution to total loss. This is a result of irreversible injury resultant from local ischemia. Figure 2-4B (about 2 days) shows a marked infiltration of the infarct by polymorphonuclear leukocytes (PMNs neutrophils). This is characteristic of the acute inflammatory response to tissue injury. By about 2 weeks (Figure 2-4C) most necrotic debris has been removed from the site of the infarct by mononuclear cells, the presence of granulation tissue, consisting of fibroblasts, newly synthesized collagen, and sprouting capillaries, indicates a transition between chronic inflammation and repair. By 5 months, the area of the infarct has been replaced by fibrosis (scaring), an acellular patch consisting of compacted collagen (Figure 2-4D). Mr. Doe has survived his myocardial infarct but has been left with a compromised heart in which a collagenous scar has replaced the normal cardiac myocytes. The pattern of necrosis, acute, and than chronic inflammation in response to cell necrosis followed by repair by fibrosis (scarring) is typical of injury to most organs and will be discussed below in detail. Complete restitution of function (regeneration) is limited to only exceptional circumstances where replicative cell populations suffice to repair tissue injury in the absence of injury to the framework (stromal component) of the organ. Superficial epidermal wounds are a common example.
Between age 58 and death at age 63, Mr. Doe suffered from increasing loss of cardiac function because of recurrent cardiac ischemia (and his unwillingness to consider invasive modes of therapy). By age 63, at the time of his death, My Doe had great difficulty in walking to his favorite coffee shop and required oxygen supplementation. His sudden death was likely the result of an additional major ischemic event. His scarred heart was particularly susceptible to conduction defects and ventricular arrhythmia was also likely to have played a role (see Chapter 7).
The etiology and pathogenesis is complex involving multiple genetic, nutritional, and behavioral factors summarized as progressive ischemic cardiovascular disease.
FIGURE 2-3
(A) Normal cardiac myocytes and (B) hypertrophic cardiac myocytes: Although cardiac myocytes do not divide, they compensate for the additional workload by increasing in size. The cells undergo endomitosis (increasing in ploidy without divison), resulting in large often rectangular nuclei (“boxcar nuclei”).
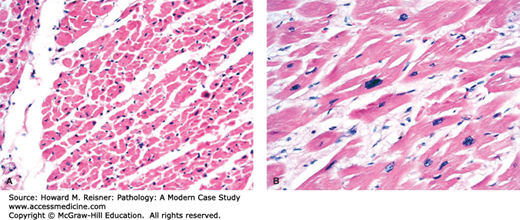
FIGURE 2-4
(A) Cardiac myocytes (12 hours postinfarct, early coagulative necrosis): Fibers are “wavy” (a likely result of mechanical stress on necrotic fibers). Some fibers are eosinophilic (arrow). Many have lost nuclei or show evidence of irreversible nuclear injury (smudging white arrow). (B) Cardiac myocytes (2 days postinfarct neutrophil infiltration acute inflammation): All fibers show evidence of necrosis and lack nuclei. Infiltration of neutrophils into tissue from vasculature is evident (arrow). (C) Cardiac myocytes (2 weeks postinfarct granulation tissue formation): Necrotic myocytes are partially removed and replaced with granulation tissue (arrow) consisting of fibroblasts, newly synthesized collagen, sprouting capillaries, and mononuclear cells. (D) Residual cardiac myocytes and scar (5 months postinfarct fibrosis): Essentially all cardiac myocytes are replaced by acellular collagenous scar. Scant mononuclear cells remain.
At the tissue level, the pathologist recognizes signs of cellular stress and injury that are potentially reversible to some degree. These patterns are often considered as normative elements of the response to increased physiological stress and hence may (or may not) be indicators of pathogenesis. These are defined as follows:
Hyperplasia is the increase in number of cells in an organ or tissue (which may, as a result, increase in size) (Figure 2-5).
Hypertrophy is the increase in size of cells in an organ or tissue (which hence increases in size). Hyperplasia and hypertrophy often coexist (Figure 2-3).
Atrophy is the decrease in the mass of an organ or tissue related to loss of cell number and/or cell size (Figure 2-6).
Metaplasia is the presumptively reversible change of one differentiated adult cell type to another. Most often this involves the change of one type of epithelial cell to another such as the change of ciliated mucosal epithelial cells to squamous in the bronchi of a smoker. Metaplasia can be pathogenetic and indicative of a “precancerous” change (Figure 2-7).
FIGURE 2-5
Two examples of endocrine gland hyperplasia: (A) Hyperplastic thyroid gland. Note the large number of follicular cells and small, highly active follicles as compared with (B) normal thyroid gland. (C) Hyperplastic parathyroid gland. The lobe is so packed with chief cells (principal cells) that connective tissue septae are not visible. This should be compared with (D) Normal parathyroid gland, which has a much lower density of cells and clearly visible septae.
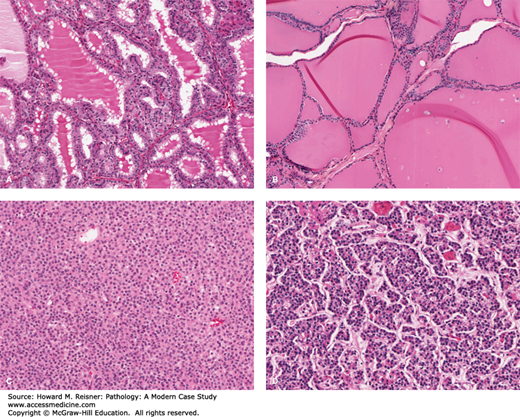
FIGURE 2-6
Denervation atrophy of skeletal muscle: (A) Denervation of skeletal muscle leads to a characteristic form of atrophy. Myofibers loose mass and the contractile apparatus becomes disorganized until little is left but residual nuclei. The atrophic fibers become angulated on cross section. Cell necrosis ultimately occurs and the myofibers are replaced by connective tissue. (B) Normal skeletal myofibers.
Another cellular adaption that, unlike those above, is clearly associated with disease is dysplasia, an abnormal change in the size, shape, and pattern of growth of cells. It can be thought of as an abnormal (or atypical) form of hyperplasia/hypertrophy and as a precursor lesion to neoplasia (see Chapter 1). Dysplasia is often associated with (and often follows) metaplasia. Hence, squamous cell cancer of the lung may begin with the above-mentioned metaplasia of the bronchus (Figure 2-8).
FIGURE 2-8
Dysplasia (cervix): This example of severe full thickness dysplasia is taken from the transition zone of the cervix (an area demonstrating squamous metaplasia). Cells have marked abnormality in size and shape. Nuclei are pleomorphic (differing in size and shape) and demonstrate abnormal mitoses.

The cells of some tissues can proliferate and/or enlarge to share workload (see Case 2-1). This often serves a beneficial function in the physiological adaptation of organs to stress. For example, the athlete “pumping iron” will experience hyperplasia of the exercised skeletal muscle and possibly skin callous formation (hyperplasia of epidermal/epithelial cells and hypertrophy of keratinized cells) where she grips the weights. Anoxic stress (of the nonacclimated to high altitudes) results in hyperplasia of erythrocyte marrow precursors; infection can lead to hyperplasia of bone marrow white cell precursors. Hormonal variations are associated with well-characterized hyperplastic and hypertrophic changes in the breast and uterus during menarche, the menstrual cycle, pregnancy, and lactation.
Hypertrophy and hyperplasia can also be associated with disease. Changes in hormones and other growth factors associated with aging result in hyperplasia of glands, smooth muscle, and connective tissue in “benign” prostatic hypertrophy (which is benign only in its lack of association with cancer). Human papilloma virus infection can result in impressive hypertrophy and hyperplasia of epithelial cells of the skin (warts). As noted above, (see Case 2-1) hypertension can result in ventricular hypertrophy that is initially physiological but can progresses to pathological ultimately resulting in heart failure and sudden cardiac death.
QUICK REVIEW Molecular Mechanisms of Hyperplasia and Hypertrophy
The molecular mechanisms of cardiac hypertrophy are of particular clinical significance because of their association with the most common disease among our aging population. The initial signals of physiological cardiac myocyte hypertrophy are biomechanical stress sensors involving integrin-mediated connections between sarcomeres and the extracellular matrix (ECM) and neurohormonal factors (such as adrenalin, angiotensin II, and endothelin) that serve to increase cardiac output in response to increased workload. Initially such signals result in compensatory increase in the expression of those genes involved in sarcomere functions, notably α-myosin heavy chain and cardiac α-actin. This increase in protein expression is associated with the addition of sarcomeres in parallel to each other and an increase in cardiomyocyte width to length ratio. Ongoing exposure of the heart to stress leads to cardiac injury and the resulting cellular response. With increased cardiac hypertrophy decreased force production occurs, vascularization is impaired, and changes in the ECM and deposition of collagen (fibrosis) occur. These nonadaptive responses are associated with induction of a distinctive array of genes:
Immediate early genes involved in the rapid and transient response of cells to stimuli, resulting in changes in gene expression profiles and new protein synthesis. These include the proto-oncogenes c-jun, c-fos, and c-myc.
Heat shock protein genes such as hsp70 that has both a protective role in stressed cells and aids in degrading damaged, misfolded proteins.
Fetal genes normally expressed in the developing heart. These include the genes for β-myosin heavy chain, skeletal α-actin, and natriuretic factors [atrial natriuretic factor (ANF) and brain natriuretic peptide (BNP)]. BNP is of note because of its utility as a convenient biomarker for ventricular dysfunction.
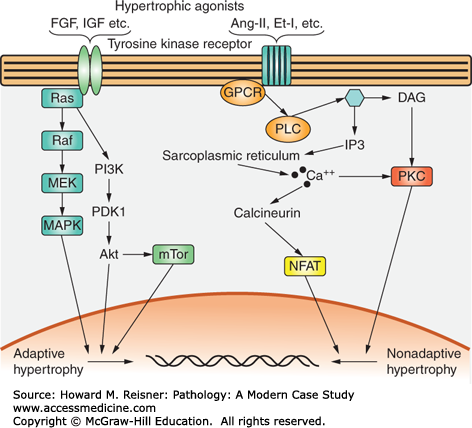
A multiplicity of signaling pathways such as ligands, cellular receptors, and intracellular signal transduction pathways are involved in the hypertrophic response. Of particular importance are the following (Figure 2-9):
Phosphoinositide 3 kinase (PI3K)/Akt pathway: Akt plays a crucial role in mediating many of the adaptive processes involved in the hypertrophic response, serving as the major effector of the PI3K signaling pathway. PI3K is activated by receptor tyrosine kinases including the insulin-like growth factor (IGF), fibroblast growth factor (FGF), and transforming growth factor (TGF) receptors. Akt inactivates glycogen synthetase kinase 3 beta (GSK-3-β) that negatively regulates cardiac size. Inactivation of this kinase leads to cardiomyocyte hypertrophy. In addition, the PI3K/Akt pathway mediates hypertrophy through the mTOR protein, a major regulator of cell growth and sensor of nutrient status. Cardiac hypertrophy is opposed by FoxO nuclear transcription factors. Akt phosphorylates and inactivates FoxO within the nucleus. The inactive phosphorylated FoxO is transported to the cytoplasm, ubiquinated and degraded in the proteasome.
G protein-coupled response (GPCR) signaling: Adrenergic factors, angiotensin II and endothelin mediate their effects via G protein-coupled receptors (GPCRs) and the Gα (q) G protein subunit and result in activation of the PI3K pathway (see above).
Calcineurin signaling: Increased Ca2+ released from the sarcoplasm in response to neurohormonal and mechanical stimuli leads to the activation of calcineurin by phosphorylation. Activated calcineurin in turn dephosphorylates nuclear factor of activated T cells (NFATs), which translocates to the nucleus where it serves to activate transcription factors important in the hypertrophic response.
Mitogen-activated protein kinase (MAPK) cascades: The MAPK pathway is a complex system of protein kinase cascades that couple external stimuli (and in particular the stress response and GPCR agonists) to nuclear responses via phosphorylation. This ultimately results in the activation of multiple transcription factors including activation transcription factor 2 (ATF-2) and myocyte enhancer factor-2 (MEF-2). Of the complex family of MAPKs, p38 kinases and c-jun NH2-terminal kinases (JNKs) are of particular importance in the pressure drive hypertrophic response.
Cytokine-mediated signaling: Members of the interleukin 6 (IL-6) family of cytokines play a role in hypertrophy by binding to the gp130 containing class of cytokine receptors followed by signal transduction using the MAPK, PI3K, and JAK/STAT. The latter pathway involves the cytoplasmic phosphorylation of STAT proteins by Janus kinases (JK). Phosphorylated STAT proteins translocate to the nucleus leading the transcription of multiple genes involved in the hypertrophic response.
The above pathways, although discussed in relation to cardiac myocyte hypertrophy, also function in the regulation of skeletal muscle hyperplasia induced by exercise. Signaling pathways important in promoting hyperplasia and hypertrophy are summarized in Figure 2-10.
Epidermal response to mechanical stress is mediated through stretch-induced Ca2++ ion channels that when triggered allow an influx of ion into the cytoplasm. This activates phospholipase C and protein kinase C and triggers the MAPK kinase pathways, ultimately resulting in a hyperplastic response. Of more general significance, the mTOR protein-mediated pathway (see above) serves a central role in controlling cell size and cell division in its role as a major nexus for cellular sensing of nutrient and energy status and by responding to systemic growth signals.
FIGURE 2-9
Factors in the development of cardiac atrophy and hypertrophy: (A) The development of cardiac atrophy involves both the inhibition of protein synthesis and a simultaneous increase in the rates of protein degradation resulting in shorter half-lives of individual cardiac proteins as compared with (B) when the half-lives of proteins are in a steady state, when protein synthesis and degradation are balanced. (C) The development of cardiac hypertrophy involves both an increased fractional synthesis of proteins and suppression of protein degradation, resulting in longer half-lives of cardiac proteins. Source: From NEJM, Willis MS, Patterson C, Proteotoxicity and Cardiac Dysfunction – Alzheimer’s Disease of the Heart?, Vol. 368, 455–464. Copyright © 2013 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
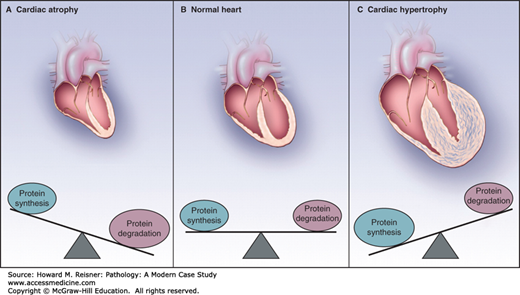
FIGURE 2-10
Major pathways activated in response to hypertrophic signals. Ang-II, angiotensin-II; DAG, diacylglycerol; Et-1, endothelin-1; FGF, fibroblast growth factor; GPCR, G protein coupled receptor; IGF, insulin-like growth factor; IP3, inositol 1,4,5-trisphosphate; MAPK, mitogen-activated protein kinase; NFAT, nuclear factor of activated T cells; PDK1; phosphoinositol-dependent kinase 1; PI3K, phosphoinositol-3-kinase; PKC, protein kinase C; PLC, phospholipase C; mTOR, mammalian target of rapamycin. Source: Reprinted from Agrawal Rohini, Neeraj Agrawal, Chintan N. Koyani, Randhir Singh. Molecular targets and regulators of cardiac hypertrophy. Pharmacological Research, Vol. 61, Issue 4, April 2010, Pages 269–280. Copyright 2010, with permission from Elsevier.
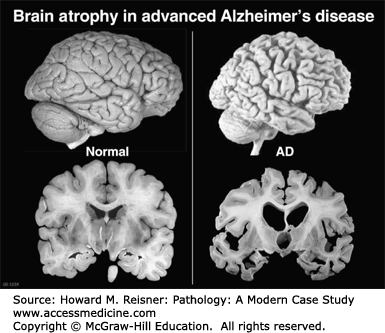
Atrophy, a decrease in organ size, is the result of a decrease in cell size (and in some cases, cell number), which like hypertrophy/hyperplasia can be either physiological or associated with disease states. Physiological atrophy is often associated with changes in hormone level (the postmenopausal breast, endometrium, vaginal epithelium, and the postparturition uterus). Aging may be associated with loss of muscle mass and “senile” atrophy of the brain and neurodegenerative disease (Figures 2-11 and 2-12).
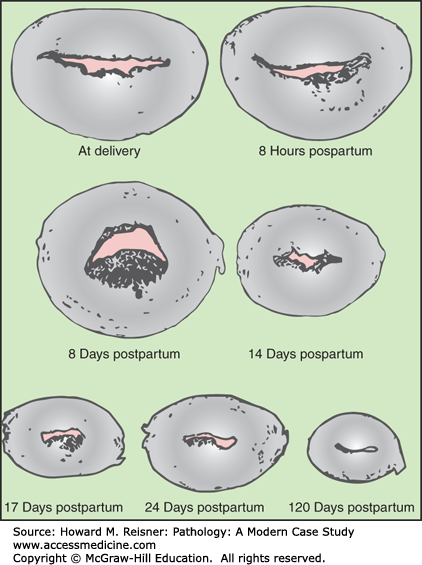
Pathological atrophy is demonstrated by muscles with decreased workload following immobilization or after loss of innervation. Cachexia, characterized by generalized weight loss, body wasting, and loss of muscle mass, is associated with several chronic diseases including cancer, renal disease, chronic obstructive pulmonary disease, and chronic heart failure. Marasmus, a result of severe malnutrition in children as a result of caloric deficit, is likewise associated with muscle atrophy and general failure of growth. Kwashiorkor, severe childhood malnutrition resulting from a diet that is very low in protein but high in carbohydrates, is both associated with growth failure and muscle wasting and also specific changes including edema, anemia changes in skin and hair pigment, excessive fat storage in the liver (fatty liver see Chapter 10), and apathy.
QUICK REVIEW Molecular Mechanisms of Atrophy
The regulation of cell (and hence organ) size requires a balance between the synthesis and breakdown of cellular proteins and other cellular components. It is not surprising that the atrophy of skeletal and cardiac muscle is associated with specific proteolytic events mediated by the ubiquitination and targeting of proteins to the 26S proteasome for degradation. Some of the key steps in the atrophic pathway can be seen as the converse of those responsible for hypertrophy/hyperplasia as follows:
Diminished systemic progrowth factors (notably IGF-1) lead to reduction in Akt activity and through additional downstream mediators, reduction in the activity of mTOR. As the central cellular sensor of pro-growth signals, mTOR plays a role in atrophy, analogous to its role in hypertrophy. Hence, lack of growth factors (or deficit in cell nutrients) leads to decreased protein synthesis and organ atrophy.
Upregulation of two muscle-specific ubiquitin ligases (ULs) occurs in the absence of Akt signaling as a result of activation of FoxO transcription factors. UL MuRF1 ubiquinates muscle myosin heavy and light chains that as a result are targeted to the proteasome for degradation, resulting in loss of sarcomere size. The UL atrogin1/MAFbx targets key protein synthesis initiation factor eIF3-f and also MyoD (a transcription factor key in muscle differentiation) for proteasomal proteolysis. Atrogin1 is a widely used marker for muscle wastage and is associated with statin-induced muscle disease. Atrogin1 also inhibits calcineurin signaling, resulting in inactivation of NFAT and loss of fetal gene expression associated with hypertrophy (see above).
Myostatin (a secreted protein member of the TGF-β family) expression is increased in skeletal and heart muscle under conditions promoting atrophy. This results in downregulation of genes important in muscle differentiation including MyoD. In addition, myostatin may promote synthesis of atrogin-1 by activating FoxO.
Cytokines including TNFa, and IL-6 and glucocorticoids enhance the ubiquitin-proteasomal degradative pathway by upregulating the NF-κB transcription factor. In this regard, the inflammatory cytokine tumor necrosis factor-like weak inducer of apoptosis (TWEAK) signals through a cellular receptor (Fn-14) that is a member of the TNF receptor superfamily. TWEAK has an important role in mediating muscle atrophy, notably in conditions of denervation and immobilization. Under such conditions, TWEAK promotes MuRF1 expression leading to muscle proteolysis.
The physiological conditions that result in atrophy, most notably aging and lack of nutrients, result in an increase in recycling of cellular components via autophagy; the process of “self-eating”, a lysosomal-dependent degradative process. In macroautophagy, organelles and portions of cytoplasm become enclosed by double membrane-bound autophagosomes that fuse with lysosomes to form autophagolysosomes (also termed “late” autophagic vacuoles). Some proteins contain a specific targeting sequence (KFERQ) that is recognized by a member of the Hsp70 family of heat shock proteins (Hsc70), which act as a lysosomal-targeting molecular chaperone (Chaperone-mediated autophagy).
As discussed in the quick review of hyperplasia (above), diminished pro-growth signaling through mTOR results in the upregulation of FoxO-dependent genes. These include several autophagy-related proteins including atg6 (Beclin-1), atg8, and atg12 that are required for autophagosome membrane formation. Starvation and the molecular mechanisms associated with it, prolong longevity in organisms as diverse as Drosophila and mammals as well as promoting atrophy and autophagy. Hence, it has been hypothesized that autophagy, by removing damaged cellular constituents and allowing recycling of limiting nutrients, may also be critical for prolongation of life (see Aging and Nutrition, this chapter).
Under conditions demonstrating significant atrophy, that is, aging and in certain neurodegenerative diseases, cells accumulate a distinctive pigment termed lipofuscin, ceroid, or “wear and tear” pigment. The material forms within autophagolysosomes and represents oxidized and cross-linked proteins and lipid peroxidation products (derived, in part, from damaged mitochondria) that are resistant to further lysosomal degradation. Using standard H&E-stained tissue, lipofuscin appears as yellow to brown granules in a perinuclear distribution (although it is more commonly detected using its autofluorescent properties) (Figure 2-13).
Metaplasia is a reversible change in which one adult cell type is replaced by another as a response to stress. Areas demonstrating metaplasia are often those in which neoplasia occurs, presumably because continuing stress is associated with loss of protective mechanisms and promotion of genetic instability. If the stressor is removed, the metaplastic cell population can return to its original state, although restoration of the initial tissue type is often incomplete. For example, the intestinal columnar metaplasia seen in Barrett esophagus as a result of chronic GERD (see Case 2-2) can completely revert to squamous epithelium that appears identical to normal following therapy when only limited areas of the esophagus are affected.
CASE 2-2
John Smith is a 47-year-old obese man who has had a history of heartburn for the past 12 years. He states that about 8 years ago he visited a gastroenterologist complaining of burning pain in his epigastric region, particularly when recumbent. Endoscopy at that time showed mild mucosal inflammatory changes consistent with gastroesophageal reflux disease (GERD) with no evidence of metaplastic or dysplastic change as confirmed by biopsy. The physician prescribed Prilosec 20 mg twice a day and elevation of the head of his bed. Five years prior to this visit, he noted increasing symptoms of reflux not always controlled by medication. He reported symptoms to be related to diet and alcohol consumption. At that time he did not complain of weight loss, dysphagia (difficulty in swallowing), or abdominal pain. Repeat endoscopy demonstrated mucosal changes consistent with Barrett esophagus, that is, the occurrence of metaplastic change within the esophagus related to GERD. Biopsy results disclosed normal esophagus (see Figure 2-14) and regions demonstrating significant metaplasia, definite low-grade dysplasia, and one localized area of concern for high-grade dysplasia (see Figure 2-15A). Because of the increased risk of developing adenocarcinoma, areas of metaplasia/dysplasia were treated by endoscopic radio frequency ablation. Mr Smith was advised to return in 3 months for potential additional therapy. At that time, he was scheduled to undergo periodic endoscopic surveillance but did not return to the clinic.
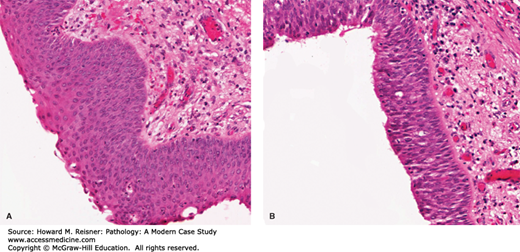
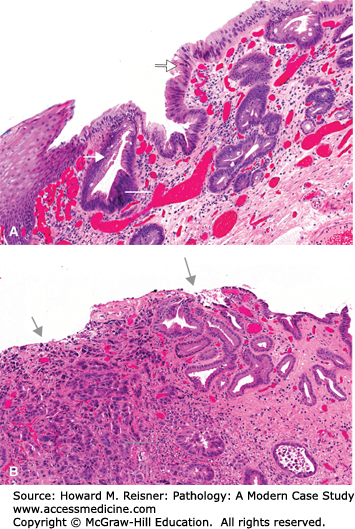
Figure 2-14 demonstrates a normal area of Mr. Smiths esophageal biopsy. The mucosa is lined with nonkeratinized squamous epithelium. The dark staining basal cells show occasional mitoses and subtend about 15–20% of the epithelial layer. Connective tissue papillae extend into the epithelial layer (arrow) and typical mucosal glands are evident (white arrow). The tissue shows hyperemia of blood vessels, likely a result of biopsy trauma. Figure 2-15A is taken from an area of the biopsy demonstrating metaplasia. The normal squamous epithelium (left margin of figure) is replaced by specialized columnar epithelium with scattered goblet cells similar to that seen in the intestine (at arrow). Other areas show cardiac type mucosa similar to that observed in the gastric cardia (at short arrow). Areas of the biopsy show evidence of dysplasia with hyperchromatic-stacked nuclei both in surface mucosa and within glands (at line) (Figures 2-14 and 2-15).
Mr. Smith now returns to the clinic with complaint of increasing dysphagia, hoarseness, and 15 pound weight loss over the last month. He states that his symptoms of GERD had worsened and are only poorly controlled by medication. Endoscopy discloses a flat lesion in the lower quarter of the esophagus adjacent to an area of Barrett metaplasia. Evaluation of biopsy discloses poorly differentiated adenocarcinoma (see Figure 2-15B short arrow) invading through the submucosa adjacent to an area showing metaplasia (Figure 2-15B arrow).
The great majority of esophageal adenocarcinomas occur in individuals with Barrett esophagus subsequent to GERD. An area of metaplastic/dysplastic change adjacent to adenocarcinoma is a common finding.
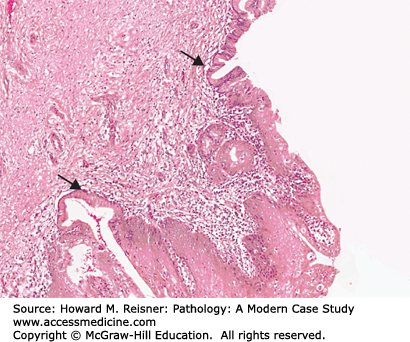
FIGURE 2-14
Region of normal esophagus: The mucosa is lined with nonkeratinized squamous epithelium. The dark staining basal cells show occasional mitoses and subtend about 15–20% of the epithelial layer. Connective tissue papillae extend into the epithelial layer (arrow), and typical mucosal glands are evident (short arrow). The tissue shows hyperemia of blood vessels, which is likely a result of biopsy trauma.
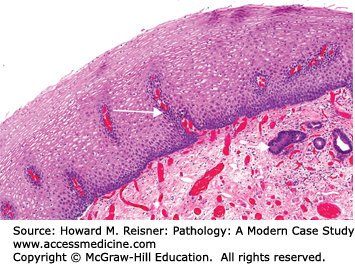
FIGURE 2-15
(A) Region of esophagus demonstrating Barrett esophagus: The normal squamous epithelium (left margin of figure) is replaced by specialized columnar epithelium with scattered goblet cells similar to that seen in the intestine (arrow). Other areas show cardiac type mucosa similar to that observed in the gastric cardia (white arrow). Areas of the biopsy show evidence of dysplasia with hyperchromatic-stacked nuclei both in surface mucosa and within glands (line). (B) Region of esophagus demonstrating adenocarcinoma: The biopsy specimen discloses poorly differentiated adenocarcinoma (short arrow) invading through the submucosa (line) adjacent to an area showing metaplasia (arrow).
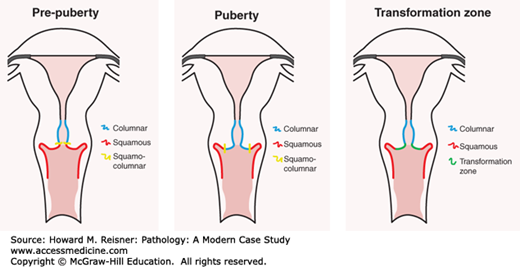
Another common example of metaplasia is seen in the change of ciliated columnar epithelium to squamous in the bronchus of a smoker demonstrated in Figure 2-7. The stressor is clearly a pyrolysis product contained in inhaled tobacco smoke, resulting in chemical injury to the cell. A third example can be found in the cervix where mucinous columnar epithelium frequently changes to squamous at the junction between the two types of epithelia (transformation zone of the cervix) (see Figure 2-16). The transformation zone of the cervix is located above the cervical os in prepubertal females. At puberty, the transition zone everts toward the exterior face of the cervix where it is exposed to the acid environment and potentially other proinflammatory agents found in the vaginal vault. In both cases (as well as in Barrett esophagus), the area of metaplasia is associated with chronic inflammation and has an increased propensity to develop cancer (Figure 2-17).
Although less commonly recognized, unexpected mesenchymal elements may form in connective tissue following injury, for example, bone may form in damaged muscle (termed myositis ossificans). The relationship of this to epithelial metaplasia is unclear.
QUICK REVIEW Molecular Mechanisms of Metaplasia
The striking association between metaplasia and the development of adenocarcinoma in Barrett esophagus has spurred study of the molecular changes that occur in the latter disease process. There is a 30-125-fold increase in risk of adenocarcinoma in patients diagnosed with Barrett metaplasia. This represents a 1 in 20 lifetime risk. The metaplasia in Barrett will progress to cancer at a rate of 0.5–1% per year.
The cell type in which Barrett metaplasia originates is in dispute. Stratified squamous epithelium might directly give rise to columnar, a process termed transdifferentiation. Such occurs during normal development of the murine esophagus in which squamous cells arise directly from columnar basal epithelial cells without intervening cell division. During this process, individual cells share cellular markers (cytokeratins) characteristic of both pathways of cellular differentiation. A more likely alternative is that an esophageal stem cell changes its developmental program to give rise to cells committed to the intestinal pathway of differentiation. Such stem cells may reside either in the esophageal mucosal interpapillary basal zone or derive from cuboidal epithelium lining the ducts of submucosal glands. Evidence gathered from patient biopsy material supports the latter source.
The strong association between GERD and Barrett metaplasia suggests that refluxed bile salts, stomach acid, and the chronic inflammation they produce are important in the etiology of the disease. The nuclear transcription factor NF-kB, key in the regulation of inflammation, is (not unexpectedly) found in increased levels in Barrett metaplasia. Two caudal-related homeobox transcription factor genes, Cdx1 and Cdx2, important in the development and maintenance of the intestine, are expressed in the normal adult gut from the duodenum through the colon but not in the esophagus or stomach. However, Cdx1 and 2 are overexpressed in areas of metaplasia in Barrett disease. Inflammation can stimulate the expression of the two genes through NF-κB, suggesting a causative link between inflammation and metaplasia induced by the ectopic expression of genes important in maintenance of adult organ patterning.
Dysplasia is not generally considered a normative response to cell and tissue injury. At the light microscopic level, it is characterized by a disordered pattern of growth in which cells show variation in size and shape and nuclear changes (Figures 2-8 and 2-15). As noted in Case 2-2, dysplasia is often found in epithelia showing metaplasia. At a molecular level, dysplasia is associated with an increasing number of mutations in genes associated with the control of cellular growth and DNA integrity, the same genetic changes that characterize neoplastic transformation. The line between neoplasia and dysplasia (if any) is exceedingly thin. In fact, what a pathologist would have called mild dysplasia of the cervix several years ago could now be called cervical intraepithelial neoplasia (CIN) grade 1. Dysplasia represents another (possibly reversible) step in the continuum of molecular changes that result in frank neoplasia and ultimately cancer.
As noted under atrophy, stressed cells may demonstrate the accumulation of abnormal amounts of substances produced locally or at other sites (Figure 2-13). The above-mentioned lipofuscin (ceroid) pigment represents residual nondegradable material predominantly within the autophagolysosomes of atrophic or aging cells. The accumulations may, in themselves, be harmless but (as is the case with lipofuscin) be indicative of a process of pathological significance. Several other common examples are as follows:
Fatty change in the liver is most often associated with excess alcohol (ethanol) consumption (alcoholic steatosis) where it represents the excess deposition of triglycerides because of both increased synthesis and reduced release as a result of disordered hepatocyte metabolism. The fat accumulation is not, of itself, damaging and is completely reversible with decreased alcohol intake. However, alcoholic steatosis may become associated with the serious consequences of alcoholic liver disease with continuing consumption.
Anthracotic pigment in the lung represents the accumulation of inhaled carbon particles in the lung alveolar macrophages. The presence of such pigment is universal in smokers as well as those living in urban environments or other areas with high levels of particulate air pollution. Carbon accumulation is not harmful but other agents found in combination with carbon (such as silica from anthracite coal production) can cause severe fibrotic lung disease.
Hemosiderosis is an abnormal intracellular accumulation of hemosiderin (a breakdown product of ferritin, the major iron storage protein in the body). The accumulation may result from genetic defects in iron metabolism or, excess hemoglobin-derived iron, as a result of hemorrhage, red cell lysis, or multiple transfusions. Excess iron may result in cellular injury via iron-mediated free radical generation (see below) (Figure 2-18).
FIGURE 2-18
Abnormal intracellular accumulations: (A) Fatty change in liver (alcoholic steatosis): Tissue processed for H&E staining (lipid extracted). (Inset): Frozen section stained for fat. (B) Anthracotic pigment (carbon) in lung. (C) Hemosiderosis in lung. Note granular brown pigment in cells. (Inset): Iron-specific stain (Perls stain). Specimen is from a case of congenital hemochromatosis.
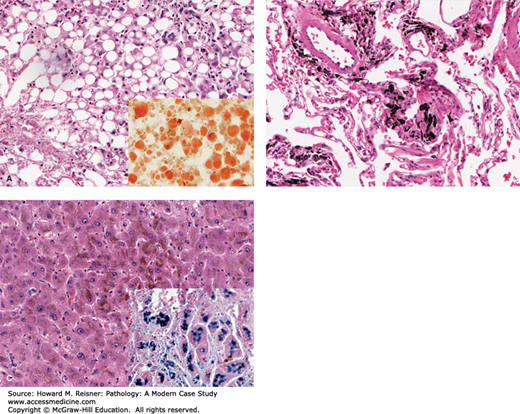
Many additional abnormal intracellular accumulations of metabolically derived or exogenous substances occur some of which are discussed in the subsequent chapters.
MECHANISMS OF CELL INJURY: NECROSIS
Although there is no clear-cut point at which cellular adaptations fail and cell injury becomes irreversible, the molecular mechanisms involved in cell injury and the histologically detectable cellular changes associated with it are well defined. Essentially all injurious agents act by disrupting one of four cellular pathways:
Aerobic respiration, mitochondrial oxidative phosphorylation, and the production of ATP,
Protein synthesis,
Maintenance of cell membrane integrity
Maintenance of the integrity of the genome (including DNA replication and repair).
Obviously, the four are interrelated; failure of any one pathway will ultimately lead to failure of the others. Of these, the first two pathways are associated with potentially reversible injury. For example, temporary impairment of blood flow to the kidneys (such as caused by a sudden decrease in blood pressure due to trauma and hemorrhage) will reduce perfusion to the renal tubules and result in local ischemia. Renal tubular cells depend, in large part, on oxidative metabolism. Much of the ATP produced is required to permit normal functioning of the Na+, K+ ATPase, the transmembrane ion pump responsible for maintaining a low intracellular Na+ concentration (and a high intracellular K+ concentration). With loss of ATP generation, the Na+, K+ ion pump fails, resulting in an influx of Na+ and water into the tubular epithelial cells causing cell swelling. Such swelling is reversible given rapid restoration of perfusion and maintenance of an intact cytoskeleton. Hence, cell swelling (sometimes called hydropic swelling) is a visible sign of reversible injury that may be seen in many organs. However, loss of or damage to the nucleus (pyknosis, nuclear condensation; karyorrhexis, nuclear fragmentation or karyolysis, nuclear loss) is considered as a sign of irreversible cell injury and, for the last, cell death (Figure 2-19).
As noted in the myocardial infarct example presented in Case 2-1, tissue ischemia is an extremely common form of cell injury associated with cardiovascular disease. Essentially every organ system is susceptible to ischemic injury, resulting from impaired circulation. Hence, the mechanisms of ischemic injury are of clinical significance and have been extensively investigated. Ischemic cell death is the model for the process termed necrosis. Necrosis is defined as cell and tissue death in an otherwise viable organism that occurs as a result of an “accidental” injury to the cell [examples being ischemia, environmental insults (such as heat, cold or physical force) and cell membrane destruction by chemical or biological agents]. Necrotic cell death is associated with early loss of internal and external membrane integrity, and release of autolytic lysosomal enzymes. Leakage of cellular contents to the tissue environment triggers multiple danger-associated molecular pattern (DAMP) sensing systems, notably pattern (or pathogen) recognition receptors (PRRs) and provokes the host response broadly termed inflammation, which will be discussed in detail in subsequent sections.
In contrast to necrosis is the process of apoptosis, or programmed cell death. Apoptosis depends on a program of complex molecular processes within the cell initiated by specific molecular triggers. Triggering of apoptosis results in a distinctive mode of cell death that is, in part, designed to minimize the host response.
Although often presented as mutually exclusive processes, mechanisms of cell death interact and overlap. A number of terms are used to better account for the complexity. For example, necroptosis is best understood as a form of receptor-mediated necrosis that shares many of the lethal mechanisms of necrosis but unlike necrosis is triggered by cytokine/receptor interaction (notably tumor necrosis factor receptor 1(TNFR1) and TNF. Pyroptosis is a form of cell death most commonly triggered by microbial pathogens but also associated with endogenous DAMPs. Like necrosis, pyroptosis results in cell swelling, lysis, and stimulation of the inflammatory reaction but unlike necrosis, pyroptosis depends on the activation of the protein caspase 1 by cytoplasmic PRRs termed “nod-like receptors” (NLRs) that will be discussed later. Activated caspase 1 forms plasma membranes pores leading to cell lysis.
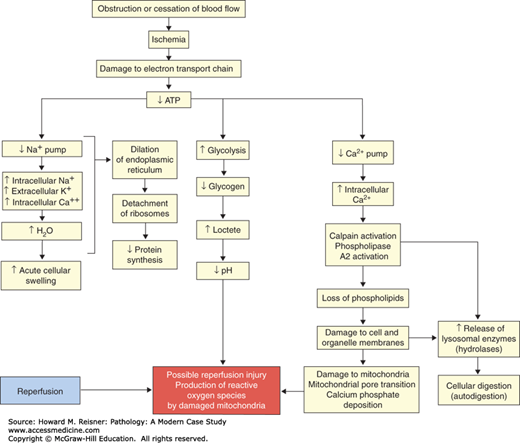
It has been suggested that a better term for necrosis might be oncosis (derived from the Greek word for swelling). Cell swelling resulting from failure of ATP-dependent ion pumps is one of the earliest evidences of ischemic cell injury. Although many critical cellular systems are deranged by ischemia, the inability of metabolically active cells to produce sufficient ATP by glycolysis alone suggests that failure of mitochondrial oxidative phosphorylation is central to the process of necrosis. For example, about 25% of ATP produced in renal tubular cells is used by the Na+, K+ ATPase ion pump. Complete renal ischemia reduces ATP levels by 70–90% in 10 minutes, most notably in the cortex. The brain, which accounts for 2% of body mass, consumes about 20% of inspired oxygen. About 70% of the energy produced in the brain is consumed by the Na+, K+ ion pump. To compensate for decreasing ATP under ischemic conditions, cells deplete glucose and stored glycogen via relatively inefficient anaerobic glycolysis, which leads to a lactic acidosis in tissue and a drop in cellular pH. Derangement of oxidative phosphorylation leads to a cascade of interacting metabolic alterations that ultimately result in cell death. These include the following (Figure 2-20):
Damage to the electron transport chain: Ischemia-induced damage to mitochondrial components leads to hydrolysis of ATP and increases in free phosphate. This serves to further reduce energy for ion pump activity.
Increased intracellular Ca2+: Under physiological conditions, there is a 10,000-fold excess in calcium concentration external to cells. Failure of the plasma membrane Ca2+ ATPase (PMCA, the major Ca2+ ion efflux pump in nonexcitable cells) results in increased intracellular levels of Ca2+. Increased cytoplasmic Ca2+ activates a number of effectors of necrosis including calpains and phospholipases and has a critical role in promoting mitochondrial damage.
Calpain activation: Calpains are Ca2+ requiring neutral cysteine proteases that have a broad range of intracellular targets. Cleavage of actin-associated proteins (including talin, vinculin, and α-actinin) can lead to membrane damage associated with cell swelling and cytoplasmic blebbing, both characteristic of necrosis. Calpains are a major factor in the disruption of lysosomal membranes leading to the release of cathepsins a family of degradative proteases normally active within the acidic environment of the lysosome, potentially a terminal event in necrosis leading to cell lysis.
Phopholipase activation: Phospholipases hydrolyze phospholipids thereby affecting the stability of cell and organelle membranes. The hydrolytic products (free fatty acids and lysophopholipids) may also damage membranes through a detergent-like activity. Many phospholipases including cytoplasmic phospholipase A2 are activated by Ca2+.
Mitochondrial damage: Increased cellular levels of Ca2+ result in increasing levels of mitochondrial calcium. Although initially tolerated, excess levels will lead to mitochondrial injury mediated by a process termed mitochondrial permeability transition (MPT). During MPT, the mitochondrial inner membrane forms pores leading to mitochondrial depolarization, uncoupling of electron transport, failure of ATP production, mitochondrial swelling (often with the presence of electron dense deposits of calcium phosphate), and lysis.
Reperfusion injury: Contraintuitively, cells that are relatively resistant to ischemic injury (such as cardiac myocytes) suffer additional injury and necrosis upon resumption of perfusion (reperfusion injury). In such cells, residual ATP (produced by glycolysis) is used by the mitochondrial ATP synthetase acting in reverse as an ATPase to allow damaged mitochondria to maintain polarization and resume function when perfusion recommences. The low intracellular pH resulting from glycolysis stabilizes mitochondria and retards MPT. Upon reperfusion such mitochondria continue to accumulate Ca2+ and, significantly, release elevated amounts of reactive oxygen species (ROS) into the cytoplasm.
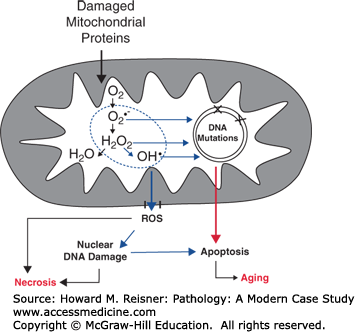
Release of ROS: Normal mitochondria leak small amounts of molecular oxygen in the form of superoxide free radical (O2•–) as a by-product of electron transport. Blocks in the respiratory chain (as would be expected in damaged mitochondria) can lead to increased production of this ROS, most notably in reperfusion injured cells. Cells have a variety of protective antioxidants to cope with ROS, but a lack of balance between protective mechanisms and ROS production leads to oxidative stress within the cell. Superoxide is rapidly dismutated to hydrogen peroxide (H2O2) by a manganese superoxide dismutase (MnSOD) located within the mitochondrial matrix. H2O2 is enzymatically detoxified to water in the presence of reduced glutathione. However, unlike superoxide, H2O2 readily diffuses through lipid membranes into the cytoplasmic space. Although not particularly reactive, H2O2 is readily converted to the highly reactive hydroxyl radical (HO•) either by reaction with superoxide (the Haber–Weiss reaction) or in the presence of ferrous iron (Fe2+) (the Fenton reaction). Lysosomes contain abundant Fe2+, are unable to detoxify H2O2, and are the major source of HO•, an effective lipid peroxidant resulting in lysosomal membrane destruction and further damage to cells. Highly reactive nitrogen compounds (such as the peroxynitrite anion ONOO–) are also generated within the mitochondria and contribute to oxidative damage. Damage may be proximate to the injury and ultimately result in necrosis. Sublethal injuries may be cumulative and play a role in aging (see Factors in Aging and their Targets, this chapter) (Figure 2-21).
FIGURE 2-21
Reactive oxygen species and mitochondrial damage. Source: Modified from Loeb, LA. The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc. Nat Acad Sci (USA) 102:18769–18770. Copyright (2005) National Academy of Sciences, U.S.A
Although the proximate cause of necrotic cell death resulting from ischemia is clearly tissue hypoxia, the subsequent cascade of metabolic failure resulting in cellular death is complex. Rupture of lysosomes, destruction of the cell membrane, or irreparable damage to mitochondria is terminal outcome but the relative roles of Ca2+ increase, ROS and failure of ATP generation by oxidative phosphorylation depend on the tissue and extent of the injury.
CELL INJURY AND NECROSIS UNDER THE MICROSCOPE
Given all the ways cells and tissues can be injured [the etiological agents of disease are immortalized in the mnemonic VINDICATE (Vascular, Immune, Neoplastic, Drug (toxicity), Infectious, Congenital, Allergic (autoimmune), Trauma, Endocrine (metabolic, nutritional)], there are many visual clues that suggest cellular injury (which may be reversible) or necrotic cell death (irreversibly injured cells extant in a living organism). Examples can include vascular injury resulting in tissue ischemia, or the effect of toxic agents (aminoglycoside antibiotics as in Case 2-3).
CASE 2-3
A 22-year-old woman presents to the emergency room following a 3-day history of fever, dysuria (pain on urination) and right-sided flank pain. She has an elevated temperature. Significant examination findings include suprapubic tenderness and pain in the right costovertebral angle (the triangular area below the 12th rib and spinal column; the area above the right kidney). Rapid urine testing is indicative of bacteremia (confirmed to be positive for Escherichia coli). She is admitted to the hospital with suspected pyelonephritis (disease of the renal pelvis and kidney associated with an infectious process in the tubulointerstitial compartment.) Such infections are most commonly “ascending” and associated with fecal bacteria that have traveled up the urinary tract via the urethra. They are more common in females because of their short urethral length. She is treated with intravenous ampicillin and gentamicin and is discharged after 4 days on continued home IV therapy.
Two Weeks LaterShe returns for follow-up and denies any continued complaints. Her physical examination is normal. However, urinalysis shows dark brown casts (material expressed from the renal tubules by the urinary stream, which contain cells and debris held together by protein secreted by the tubular cells). Her blood urea nitrogen (BUN) and serum creatinine are both elevated. Such results are suggestive of nephrotoxic tubular injury consistent with aminoglycoside antibiotic therapy-induced renal toxicity.
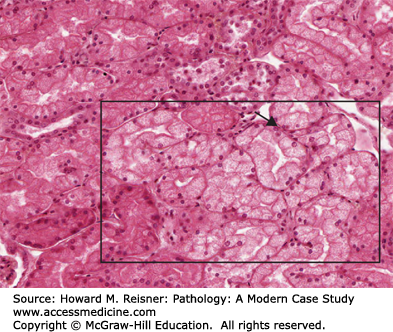
Although it is unlikely that a renal biopsy would be ordered, this is a well-understood disease process Figure 2-22 is consistent with the patient’s disease.
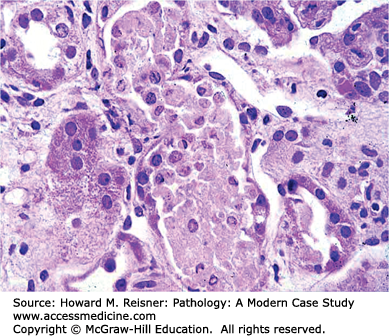
Visual clues associated with reversible injury are cellular swelling (so-called hydropic swelling), an increase in intracellular swelling associated with failure of the ATP-dependent Na/K ion pump. In addition, plasma membrane blebbing, disaggregation of ribosomes and dilation of the endoplasmic reticulum, aggregation of cytoskeletal elements, and variable changes in mitochondria such as swelling and formation of internal calcium phosphate precipitates may variably occur. Irreversible injury is histologically defined by loss or disruption of the nucleus. Hence, nuclear loss or disruption of cells observed microscopically is a useful feature indicating necrosis (as opposed to reversible injury). Changes such as increased cytoplasmic staining with eosin (likely resulting from loss of mRNA), defects in the cell membrane, the formation of large densities in mitochondria, and rupture of lysosomes can be difficult to evaluate at the light microscopy level. Of course, gross enzymatic digestion of cells or tissue (autolysis when a product of the cell’s own enzymes, as might be seen in postmortem tissue samples, heterolysis if the enzymes are produced by another source, as in certain tissue destructive bacterial infections) is unequivocal. Figure 2-24 provides examples drawn from clinical material.
FIGURE 2-24
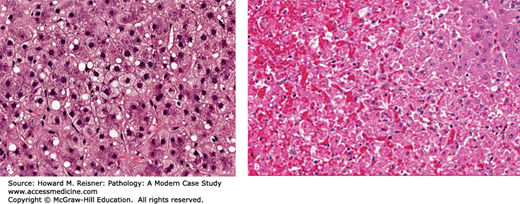
Reversible and irreversible injury. Two examples from the liver: (A) Potentially reversible cell swelling in a liver damaged by ethanol consumption. (B) Irreversible injury in a neonatal liver infected with herpes simplex. Hepatocytes have undergone varying degrees of nuclear damage and in many cells total loss. Some hemorrhage is evident.
As noted above, pathologists recognize patterns of necrosis that may be tissue specific or provide suggestions as to the etiology of injury. The names associated with such patterns although “traditional” are descriptive and useful.
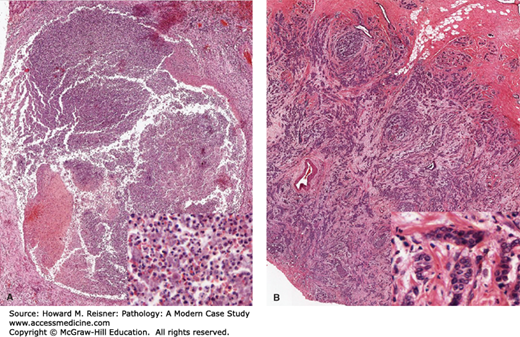
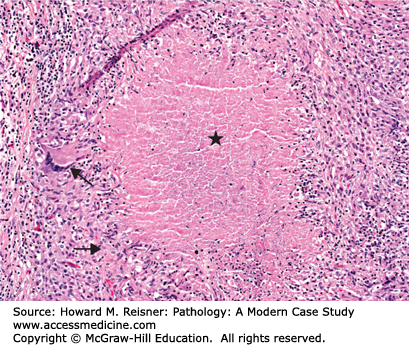
Coagulative necrosis: This form of necrosis occurs when enzymatic digestion/liquefaction of tissue by autolysis is retarded and coagulation (protein denaturation) predominates. The remnants of cells remain in place in tissue for some time as a necrotic area containing “cell ghosts.” Lack of nuclei and nuclear disruption is obvious. Areas may show eosinophilia (as long as cytoplasmic proteins remain intact) or simply show reduced overall intensity of staining. This pattern of necrosis is commonly associated with vascular lesions that produce tissue ischemia and not only result in infarcts as in the heart (myocardial infarct; “heart attack”) but also in other organs (kidney and spleen being common sites) (Figure 2-25).
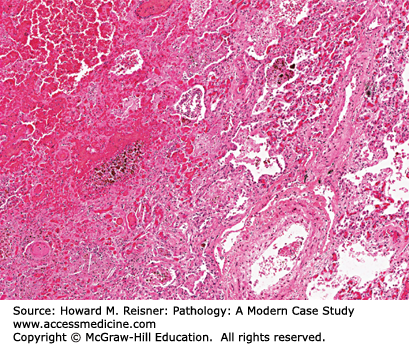
Coagulative necrosis and hemorrhage and (incorrectly hemorrhagic necrosis): Infarcts in organs that have dual blood supplies such as the lung and occasionally the liver may result in coagulative necrosis in the presence of sufficient residual circulation (from an alternate source) so as to demonstrate massive tissue hemorrhage. This is best demonstrated in the lung where pulmonary infarcts resulting from pulmonary embolic disease (PEs in clinical jargon) are a common problem, resulting in significant mortality (Figure 2-26).
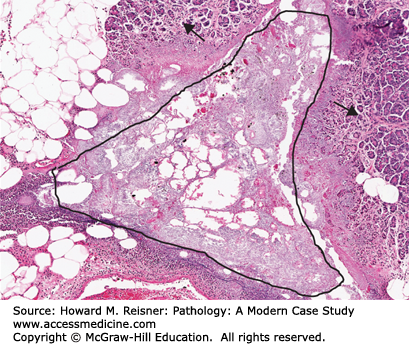
Liquefactive necrosis: This form of necrosis commonly occurs in abscesses resulting from a bacterial infection where cellular enzymes are released from polymorphonuclear neutrophils (PMN) reacting as part of the acute inflammatory response release hydrolytic and other enzymes, resulting in tissue liquefaction. The resultant mixture of cells and debris is termed pus. Liquefactive necrosis also occurs in neural tissue in the case of infarcts. This ultimately results in a fluid-filled cyst (e.g., in the brain following an ischemic event, a stroke) (Figure 2-27).
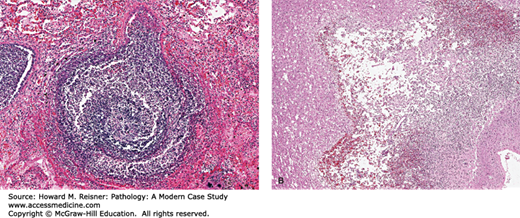
(Enzymatic) fat necrosis: As mentioned above, this process occurs in tissues with many adipocytes. Destruction of such cells releases triglycerides. Lipases (also released from damaged cells, particularly pancreatic acinar cells) convert the triglycerides into free fatty acids, which combine with tissue calcium to form soaps. The soaps are basic, highly destructive, and irritating. Enzymatic fat necrosis is a serious and potentially life-threatening complication of pancreatitis and surgical or other accidents damaging the pancreas and its exocrine ducts (Figure 2-28).
Caseous necrosis: As briefly discussed above, this form of necrosis is characteristic of the host response to infections with tuberculosis and will be discussed later. In this response, compact aggregates of inflammatory cells (primarily macrophages termed epithelioid cells) called granulomas have central areas of characteristic “cheesy” necrosis, hence are termed caseating granulomas (Figure 2-29).
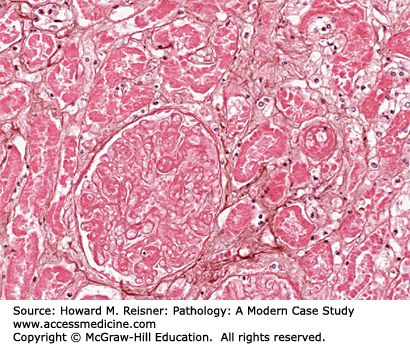
FIGURE 2-26
Coagulative necrosis and hemorrhage: The result of a recent pulmonary infarct that resulted in coagulative necrosis of alveoli and subsequent hemorrhage from extant circulation. The left side of the image is hemorrhagic with some anthracotic pigment visible (black punctate material). The right side shows necrotic alveoli and vasculature.
WHAT WE DO
The ability of the pathologist to distinguish the histopathological signs of cell and tissue injury and death is of practical importance. At a minimum, it will direct attention to areas requiring further analysis. Different patterns of injury and necrosis may provide clues as to the etiology of injury and the pathogenetic process involved. For example, bacteria often produce a pattern termed liquefactive necrosis. An isolated or encapsulated area of liquefactive necrosis (an abscess) suggests a nidus of bacterial infection. Coagulative necrosis leaves the resistant structural architecture of tissue intact and is often described as “leaving in the ghostly remains of tissue intact.” It is characteristic of ischemic necrosis. Caseating granulomatous necrosis is a strong indicator of infection by Mycoplasma tuberculosis. Particular patterns of necrosis are associated with specific tissues; enzymatic fat necrosis is associated with tissue rich in fat cells, most commonly the peripancreatic area, occasionally the breast and buttock. It is important to consider that these distinctions predated our knowledge of causation; hence, terminology does not always fit the biochemical facts. (Caseating necrosis is so called because German pathologists in the 19th century thought the tissue lesion looked like crumbly cheese-Käse.) Nevertheless, the ability to visually identify these types of necrosis in tissue is of continuing clinical utility and the terms frequently appear in surgical pathology reports. Consider a 2 cm indurated lesion on a woman’s breast. On biopsy will it be an abscess demonstrating liquefactive necrosis within, or will it be a clonal proliferation of neoplastic cells, cancer? Obviously the difference is significant in terms of therapy and prognosis. Pathologists sitting at their microscopes help make the distinction (Figure 2-23).
MECHANISMS OF CELL INJURY: APOPTOSIS
Necrosis is disorderly; apoptosis is highly programmed and orderly. Necrosis results in the release of cellular components into the tissue environment where they stimulate a vascular and cellular response termed inflammation. As shall be discussed, the inflammatory response may, of itself, provoke additional tissue injury (such as abscess formation). Most often, necrosis is provoked by results external to the cell itself such as tissue ischemia, physical injury produced by heat or force injury, and in some cases bacterial products. In such cases, energy production by the cell may cease abruptly. Given the specifics of injury, cells may or may not be able to survive, for a time, using anaerobic metabolism.
Apoptosis (from the Greek for “a falling off”) is a highly programmed and orderly form of cellular suicide that may be provoked by specific external or internal cellular signals. It has been referred to as “controlled demolition at the cellular level.” Although viewed by pathologists in the context of cellular injury, apoptosis is critical for normal embryogenesis and in the adult, for hormonally dependent loss of cell mass such as regression of the lactating breast and endometrial breakdown during menses.
Importantly, apoptosis is an energy requiring process that can only occur in cells that maintain some degree of metabolic integrity. Apoptotic cells shrink, while organelles remain relatively normal. Very early in the process of apoptosis a specific DNase is activated that results in a characteristic “ladder pattern” of DNA fragments. The degraded chromatin condenses, forming aggregates. The cells show extensive surface blebbing and (with a few exceptions) proceed to fragment into membrane-bound apoptotic bodies. These are neatly packaged cellular contents that otherwise would spill into the tissue environment acting as “danger signals” triggering host defenses (and particularly innate immunity). Apoptotic bodies display the membrane phospholipid phosphatidylserine (PTS) and other molecules on their surface that act as ligands for phagocytic cells thus promoting tidy disposal. To reiterate, the key utility of the process of apoptosis is to provide cells with the ability to suicide without provoking (potentially damaging) host responses. Necrosis, on the other hand, serves to provoke such responses (Figure 2-30).
FIGURE 2-30
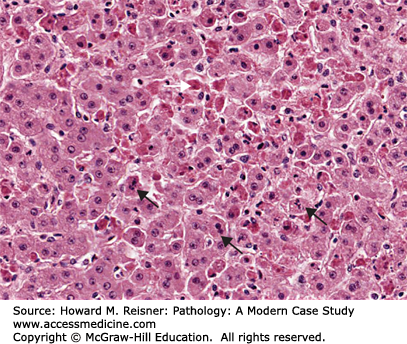
Apoptotic bodies: Hepatocytes in the liver from a case of yellow fever. Many of the cells are in various stages of apoptosis. For example, some cells (arrows) demonstrate a condensed fragmented nucleus. The eponymous term “Councilman body” refers to an anucleate eosinophilic remnant of a hepatocyte that has undergone apoptosis (apoptotic body) (white arrow).
QUICK REVIEW Molecular Mechanisms of Apoptosis
Although apoptosis is now understood in great detail, initial understanding of the system grew from a simple observation in the nematode worm Caenorhabditis elegans. The worm has an absolutely fixed pattern of development that features 1090 somatic cells, 131 of which must die for normal body formation. Three highly conserved genes regulate this process. One of the genes (CED3) is homologous to the initially characterized caspase in humans (caspase 1).
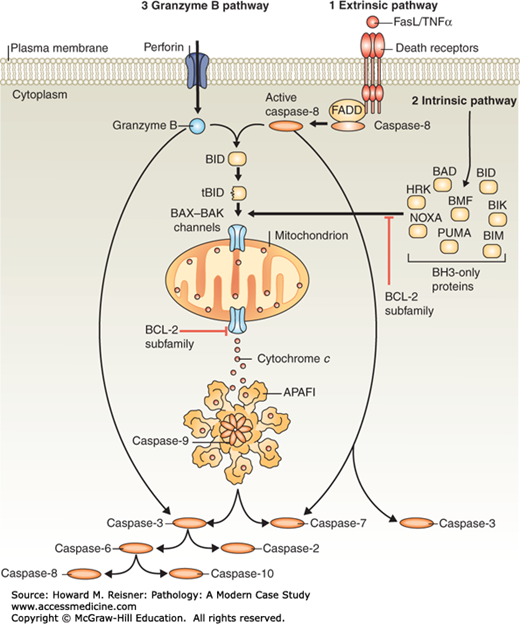
The caspases are key to apoptosis, the result of a complex series of cascading enzyme interactions. The caspases are unique proteases containing an active site cysteine, which cleaves target proteins after aspartic acid residues. Caspases normally exist in an inactive form and must be proteolytically cleaved to become active. This is often accomplished by another active caspase. Hence, a critical issue is what are the initial triggers that start the process. A reasonable approach to this is to divide apoptotic caspase activation into three pathways: (1) extrinsic, (2) intrinsic, and (3) granzyme B mediated. (The third pathway is sometimes referred to using different names such as “cytotoxic T lymphocyte (CTL) mediated.). The three pathways utilize specific initiator caspases capable of autoactivation to start the apoptotic process. All three pathways converge on the activation of the effector caspases 3, 6, and 7, which are responsible for most, if not all, of the cellular changes (demolition) mentioned above. [Parenthetically not all caspases are involved in apoptosis. Several of them (including the prototypical caspase 1) are involved in innate immunity.] (Figure 2-31).
Extrinsic Pathway
Activation commences with the engagement of an extracellular ligand by death receptors on the surface of the cell. Receptors are members of the tumor necrosis factor receptor family (TNFr) and have an intracytoplasmic death domain (DD), which couples cell surface signals to the intracellular environment. Examples of receptor/ligand pairs are Fas (CD95)/Fas ligand (CD95L) and TNF receptor 1/TNFα.
Death receptor ligand engagement results in receptor trimerization and recruitment of DD containing adaptor proteins (FADD Fas-associated death domain) to form a death-inducing signaling complex (DISC).
The DISC promotes oligomerization and autoactivation of initiator caspase 8.
Activated caspase 8 may directly activate executioner caspases such as caspase 3 or feed into (and be amplified by) the mitochondrial-dependent intrinsic pathway by cleaving a member of the Bcl-2 homology group of proteins, Bid.
Cleaved Bid relocates to the mitochondria where it triggers the intrinsic apoptotic pathway by cleaving the Bcl-2 proteins, Bak and Bax.
The entry of a cell into the intrinsic pathway depends on the balance between pro- and antiapoptotic members of the Bcl-2 family of proteins that are defined by the presence of BH homology domains.
The antiapoptotic proteins (such as Bcl-2) bind to and inhibit the polymerization of proapoptotic members of the family. An additional class of BCL-2 proteins (BH-3 only proteins) promotes apoptosis when expression levels are increased and serve to integrate a variety of cellular signals into the triggering of apoptosis. For example, levels of Noxa and Puma are upregulated by the p53 system and leading to apoptotic death of cells with irreparable DNA damage. As noted above, Bid integrates the extrinsic apoptotic pathway into the intrinsic.
Once activated, Bak and Bax cause permeabilization of the mitochondrial outer membrane (MOMP) and subsequent cytosolic release of intramembrane mitochondrial proteins, notably cytochrome c.
Released cytochrome c binds to the cytoplasmic protein Apaf-1, which together with dATP oligomerizes to serve as a scaffold for the binding and activation of the initiator caspase 9.
The cytochrome c, Apaf-1 caspase 9 complex (the apoptosome) activates executioner caspase 3 and (directly or indirectly) activates executioner caspases 6 and 7.
CTLs and NK cells mediate cell killing by inducing the formation of perforin pores in targets allowing for the entry of the caspase-like protease granzyme B.
Granzyme B can either directly activate executioner caspase 3 or cleave Bid triggering the intrinsic apoptotic pathway.
Proteolysis of cellular components by executioner caspases is either directly or indirectly responsible for the events that characterize apoptosis. For example, production of the characteristic 180 bp DNA ladder during apoptosis (an often used biochemical indicator of the process) is the result of cleavage of DNA by a specific caspase 3 activated DNase (CAD). Caspase 3 cleaves an inhibitor of CAD (iCAD) activating the nuclease. Although seen as pathophysiology by the pathologist, active caspase 3 is necessary for cell differentiation. During development, caspase 3/CAD-induced DNA strand breaks are required for normal myogenesis.
IN TRANSLATION Significance of Cell Death Scenarios
The four different mechanisms of cell death mentioned (necrosis, necroptosis, pyroptosis, and apoptosis) demonstrate several recurrent themes. The first three trigger host responses to injury (inflammation or, more broadly, innate immunity) either specifically (necroptosis and pyroptosis) or nonspecifically (necrosis) The fourth uses multiple mechanisms to prevent (and in some cases suppress) the host response.
The significance of these cell death scenarios for the future physician involves the fact that inflammation can be destructive as well as protective. Inflammation can rapidly exceed its protective role and produce cellular injury, resulting in systemic shock and multiorgan failure syndromes (systemic multiorgan response syndrome) with their significant morbidity and mortality. At the same time, anti-inflammatories are major therapeutic agents. However, many of these agents are directed at specific classes of inflammatory mediators such as arachidonic acid (AA) metabolites [predominantly prostaglandins (PGs)] or specific cytokines (such as TNFα or IL-1). Excessive stimulation of inflammation may result in a storm of interacting mediators, which no one drug, or group of drugs, can effectively suppress. Thus, understanding the initial triggering events of proinflammatory cellular processes would (at least in theory) provide a way of modulating the degree of inflammation. For example, modulation of caspase 1 activation would control pyroptosis, a critical event provoking the inflammatory reaction to a wide variety of pathogens. Hence, new classes of therapeutic agents controlling the initial events that trigger innate immunity are an area of great interest to the pharmaceutical industry.
Although physicians have an interest in being able to “turn off” or at least might wish to modulate, those modes of cell death that lead to inflammation, there is also an interest in being able to turn on apoptosis. Suppression of apoptosis by inactivation of components of the p53 pathway (e.g., by mutation of the TP53 gene) is associated with increased malignancy in many forms of cancer. The p53 system that initiates apoptosis in cells which have irrecoverable genomic damage is important both in the progression of cancer and in mediating the effects of many cytotoxic chemotherapeutic agents, which act by inducing DNA damage. Hence, cells with defective p53 function may rapidly accumulate additional somatic mutations furthering tumor growth while also becoming resistant to chemotherapy. The ability to induce apoptosis in such malignant cells is an attractive potential approach in treating malignancies currently undergoing clinical trials. Agents, which inactivate antiapoptotic proteins (such as BCL2) or mimics of proapoptotic BH3 proteins, have shown promise in a number of cancers. Cancer cells are particularly sensitive to activation of the extrinsic apoptotic pathway via the TNF-related apoptosis inducing ligand (TRAIL). The use of TRAIL or antibodies directed toward the TRAIL receptor is also being evaluated as for clinical utility.
HOST RESPONSE TO INJURY
The major threats to the human organism, that is, the etiological agents of disease and death, have remained constant during our evolutionary history. The specific array of pathogens, be they viral, bacterial, fungal or parasitic, changes with time, but their overall challenge to our survival does not. Force injury (physical or thermal) currently kills far more children in the United States than do pathogens, which are the major challenge to childhood survival in most of the underdeveloped world. Although the balance of threats alters, responding to temporal and geographic change, the nature of the threats does not. Thus, evolution provides us with an array of damage sensors that recognize characteristic molecular patterns associated with pathogens and cell injury, the pattern/pathogen recognition receptors (PRRs) that recognize DAMPs. The first result of triggering PRRs is the initiation of innate immunity, defined as host protective responses that do not involve variable region-dependent immunity. Innate immunity is preprogrammed to quickly respond to specific DAMPs, which are presumed to have presented a continuing challenge to the organism during evolution. Innate immunity includes what pathologists define as the acute inflammatory response with emphasis on triggering events. Adaptive immunity, traditionally the purview of the immunologist (and immunopathologist), deals with cellular and humoral responses that have the ability to adapt to new challenges and improve with continuing exposure. Such challenges need not have been present during evolution, but can be new to the species (such as synthetic organic molecules). Adaptive immunity tends to be slower than innate but with time the two systems interact and amplify each other. For example, chronic inflammation as defined by pathologists involves elements of both systems. The response to infectious agents most often involves such interplay.
Force injuries do not, of necessity, involve pathogens foreign to the injured host. Although the sites of injury may rapidly become infected with exogenous agents, the immediate host defense to “sterile injury” relies on the sensing of endogenous molecules (sometimes termed alarmins) released from injured cells and not normally present in the tissue environment.
High-mobility group box 1 (HMGB1) protein is a well-characterized prototypical alarmin that may be passively released from sterilely injured necrotic cells as well as actively released from immune and epithelial cells following either stress or exposure to proinflammatory cytokines (IL-1, TNFβ, IFN-γ). Actively secreted HMGB1 is important as a late acting mediator in systemic inflammation such as that resulting from severe sepsis. However, HMGB1 is also important in the early innate immune response since passively released HMGB1 binds to a number of receptors important in that response (toll-like receptors TLR-2 and -4 of myeloid cells and RAGE, discussed below). This results in activation of the NF-κB pathway and synthesis of a wide variety of inflammatory and chemotactic cytokines (such as CXCL12) from dendritic cells and monocyte/macrophages and promotes the early migration of leukocytes to the site of sterile injury. Other molecules that are predominantly released by necrotic cells and show alarmin-like stimulation of innate immunity include IL-1α, heat-shock proteins, uric acid, and hyaluronan.
During apoptosis, IL-1α is sequestered in the nucleus and not released. However, the molecule is readily released by necrotic cells and induces macrophages and neutrophils to release numerous cytokines (such as IL-6 and TNFα) and promotes accumulation of such cells at the site of injury. Released IL-1α is a major alarmin and crucial for the initiation of inflammation under such conditions.
Mitochondrial DNA and N-formyl peptides synthesized and released by damaged mitochondria also trigger PRRs (possibly because of the prokaryotic ancestry of mitochondria). Mitochondrial derived alarmins specifically trigger neutrophil PRRs and induce migration and degranulation of these cells in response to injury. Hence, mitochondrial damage is important in triggering the early cellular phase of acute inflammation (discussed below).
Pathogen-associated molecular patterns (often termed PAMPs) are molecules associated with pathogens foreign to self (with the exception of damaged mitochondrial products noted above). In general, PAMPs are essential for pathogen survival and unlikely to be lost by selection. A long and nonexhaustive list includes (1) microbial membrane components (notably lipopolysaccharide LPS, peptidoglycans, and other molecules), (2) surface glycoproteins, (3) bacterial flagella, (4) mannose containing polymers, (5) bacterial N-formylmethionyl containing peptides, and (6) nucleic acids (such as viral double-stranded and single-stranded RNA, and bacterial DNA characterized by unmethylated CpG motifs). The list of PRRs sensing PAMPs is equally long and includes cell membrane bound, intracellular, and secreted molecules specialized to deal with particular classes of molecules and pathogens. For example, intracellular PRRs deal with viral or intracellular bacterial threats. PRRs are germ line encoded and are often expressed constitutively (although upregulation may occur during pathogen challenge). The best-known set of receptors are the cell membrane located toll-like receptors (TLRs) predominantly expressed on normal macrophages and dendritic cells (but also on other specialized cell populations such as enteroendocrine cells of the intestine and in colonic epithelial cells).
Although TLRs were first characterized in Drosophila, homologous proteins exist throughout the animal kingdom where they play a role in host defense. Hence, they represent what is likely to be the earliest host defense PRR to evolve. TLRs are expressed as integral membrane glycoproteins present on either the cell surface (TLRs-1, -2, -4, -5, -6) or within intracellular vesicles such as endosomes, endoplasmic reticulum, or lysosomes (TLRs-3, -7, -8, -9). Humans have 10 functional TLRs that are type I transmembrane proteins each of which has a differing specificity based on their leucine-rich repeat (LRR) containing ectodomains. Downstream signaling is mediated via an intracellular toll interleukin-1-like receptor (TIR) domain. The downstream signaling via TIR is dependent on sets of specific signaling molecules that yield responses that differ depending on the nature of the PAMP sensed and the cell bearing the receptor. Adapter MyD88 is used by all TLRs except TLR3 and ultimately stimulates the production of proinflammatory cytokines via NF-κB and MAP kinases. Adaptor TRIF is active in TLRs-3, -4, -7, and -9 and results in downstream induction of type I interferons with antiviral activity in addition to proinflammatory cytokines stimulated via Myd88 (Figure 2-32).
FIGURE 2-32
Localization and signaling pathways of TLRs: Note TLR-2 forms functional heterodimers with TLRs-1 and -6. MAL MyD88 adaptor like proteins; TRAM, TRIF-related adaptor molecule. See text for additional details. Source: This research was originally publised in Clinical Science. Vallejo J. Role of toll-like receptors in cardiovascular disease. Clin Sci. 2011; 121:1–10, © copyright the Biochemical Society.
The PAMP specificity of particular TLRs has been clarified using murine knockouts. In general, the intracellular TLRs (3,7,8 and 9) are nucleic acid sensors detecting the products of bacterial and viral pathogens, whereas the cell surface TLRs sense a wide array of bacterial, fungal, and parasitic structural components. TLRs-1, -2, and -6 recognize bacterial-derived lipids and are important in sensing gram-positive bacteria; TLR-4 (in complexes with another protein, MD-2) recognizes the bacterial LPS of Gram-negative bacteria, which is responsible for septic shock. TLR-5 binds bacterial flagellin and is heavily expressed on epithelial cells of the gut and respiratory tree where it is important in localized host defense.
An ever-increasing set of cytoplasmic PRRs is being defined. They detect bacteria that escape into the cytoplasm (e.g., Shigella, Listeria, and invasive E. coli). These include nuclear-binding and oligomerization domain (NOD)-like receptors (NLRs) and retinoid acid-inducible gene-I (RIG-1)-like receptors (RLRs).
There are over 20 NLR genes in the human genome, which are organized into groups based on their amino-terminal protein–protein interaction domain.
NLRCs contain a caspase recruitment domain (CARD)
NLRPs have a pyrin domain (PYD)
NAIPs have a baculovirus inhibitor repeat domain (BIR)
Both CARD and PYD domains are members of the “death domain fold” superfamily so named because of their importance in mediating the formation of high molecular weight complexes via self-association that are important in caspase activation and apoptosis. The BIR is a zinc-finger domain characteristic of a number of antiapoptotic proteins. All NLRs have a characteristic NOD domain necessary for oligonucleotide binding and self-association and a C-terminal LRR region conferring PAMP specificity. Binding of PAMPs by the LRR permits the NOD regions to oligomerize and expose the effector domains allowing them to interact thereby promoting interactions with other CARD or PYD effector molecules (Figure 2-33).
FIGURE 2-33
Families of pathogen receptors, TLRs, NLRs, and RLRs:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


