Celiac Disease1
Carol E. Semrad
1Abbreviations: AGA, antigliadin antibody; DGP, deamidated gliadin peptide; EMA, endomysial antibody; HLA, human leukocyte antigen; Ig, immunoglobulin; IL, interleukin; tTG, tissue transglu taminase.
Coeliac affection was first described as a diarrheal illness related to diet by Gee in 1888 (1). In 1950, Dicke (2, 3) reported that wheat was the cause. He observed that, during the grain shortage of World War II, children with celiac disease miraculously improved in weight and stature on a potato diet, only to relapse when bread was again available after the war. In the intervening years, many advances have been made to characterize the component of wheat responsible for triggering celiac disease and the intestinal immune response. Based on this work, celiac disease is defined as a T-cell-mediated inflammatory disease of predominantly small bowel triggered by gluten in the diet from wheat, rye, and barley in persons who are genetically susceptible. The consequences of chronic small bowel inflammation are mucosal atrophy and malabsorption of macronutrients, vitamins, and minerals. This chapter focuses on the clinical presentation and pathogenesis of celiac disease, its nutritional consequences, and the gluten-free diet.
PATHOGENESIS
Epidemiology
The prevalence of celiac disease in the United States is 1 in 133 persons, based on blood donor screening using sensitive and specific antibody tests (4). The worldwide prevalence of celiac disease in whites is approximately 1% (5), and the rate of diagnosis appears to be increasing (6, 7). Proposed theories for its increasing prevalence include cultivation of wheat grains with higher gluten content, rotavirus infection that may increase intestinal permeability (8), and change in breast-feeding practices (9). Breast-feeding with the introduction of a small amount of gluten between 5 and 7 months of age may prevent or delay the onset of celiac disease in genetically susceptible infants (10). Celiac disease most commonly manifests at age 1 to 2 years, when gluten is first introduced into the diet, and in young adult life, but it can become evident at any age (11).
Pathobiology
The development of celiac disease requires the ingestion of gluten from wheat (includes spelt, triticale, semolina, and Kamut), rye (including malt), or barley (Fig. 79.1) and a susceptible genetic background (5, 6, 7). Gluten is the protein in wheat that provides the elastic quality highly desirable for baking goods. Repeat stretches of the amino acids proline and glutamine are characteristic of the gluten protein structure. The alcohol-soluble fraction of wheat gluten, gliadin, and its related prolamins from rye and barley are toxic in celiac disease (12, 13). Many gliadin peptides have been reported to trigger inflammation in celiac disease by in vivo and in vitro studies. One such peptide, a 33-mer peptide from α2-gliadin (14), contains three previously reported toxic epitopes and cannot be digested by human pancreatic or intestinal proteases, thus rendering it available to trigger an immune response.
A genetic predisposition for celiac disease is suggested by the high concordance of celiac disease in identical twins (15) and the increased risk in first-degree relatives compared with the general population (16). The human leukocyte antigen (HLA)-DQ2 (DQA1*05/DQB1*02) and DQ8 (DQA1*03/DQB1*0302) alleles have the strongest genetic association with celiac disease (17). However, not
all individuals with these risk alleles develop the disease. Other candidate genes have been identified in genomewide studies (11, 18, 19, 20) that, when present, along with HLA-DQ2, increase the risk of celiac disease (21). Genetic associations also have been reported between celiac disease and diabetes mellitus type 1(22), as well as Crohn disease (19, 23).
all individuals with these risk alleles develop the disease. Other candidate genes have been identified in genomewide studies (11, 18, 19, 20) that, when present, along with HLA-DQ2, increase the risk of celiac disease (21). Genetic associations also have been reported between celiac disease and diabetes mellitus type 1(22), as well as Crohn disease (19, 23).
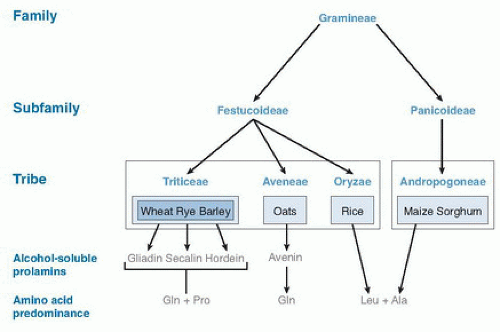 Fig. 79.1. Family of grasses. Ala, alanine; Gln, glutamine; Leu, leucine; Pro, proline. |
Gliadin has been reported to trigger both adaptive and innate immune responses in celiac intestinal mucosa (Fig. 79.2). How gliadin peptides cross the intestinal epithelium is unknown. Intestinal infection or other physiologic stress, such as surgery, may render the epithelium more permeable and allow a threshold amount of gliadin peptide access to immune cells in the lamina propria. Alternatively, peptides cross transcellularly (24, 25). In the lamina propria, native gliadin peptides or gliadin peptides rendered more negatively charged by conversion of glutamine to glutamic acid by tissue transglutaminase (tTG) bind to positively charged pockets of HLA-DQ2 and DQ8 molecules expressed on the outer membrane of antigenpresenting cells (11, 26) (see Fig. 79.2). These bound antigens activate specific T cells that result in release of interferon-γ and intestinal inflammation. Gliadin is also capable of activating the innate immune system by reprogramming intestinal epithelial cytotoxic T lymphocytes into natural killer-like cells (27). Release of the cytokine interleukin-15 (IL-15) from the epithelium participates in intestinal inflammation. Retinoic acid may act as a coadjuvant to IL-15 to break tolerance to gliadin peptides in individuals genetically predisposed to celiac disease (28).
The main pathophysiologic consequences of chronic intestinal inflammation are villous atrophy and decreased surface area for nutrient absorption. Chronic inflammation may also down-regulate nutrient transport proteins in the intestinal epithelium (29). Pancreatic insufficiency caused by decreased release of secretin and cholecystokinin from atrophied duodenal epithelium and bacterial overgrowth may contribute to nutrient malabsorption.
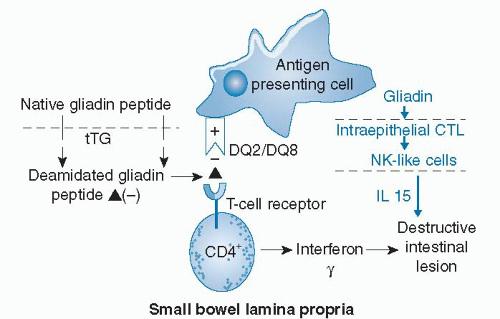 Fig. 79.2. Pathogenesis of celiac disease. Poorly digestible gliadin peptides, such as the 33-mer, cross the intestinal epithelium by an unknown mechanism and reach the lamina propria. Native peptides or peptides deamidated by tissue transglutaminase (tTG) bind to the positively charged human leukocyte antigen (HLA)-DQ2 or DQ8 molecules expressed on the outer membrane of antigen-presenting cells and are recognized by CD4+ T cells. T-cell activation results in release of interferon-γ and destruction of the epithelium. Gliadin also acts directly on the intestinal epithelium to stimulate release of interleukin-15 (IL-15) that can synergize the immune response in the lamina propria. CTL, cytotoxic lymphocyte; NK, natural killer. (Adapted with permission from GreenPH, Jabri B. Coeliac disease. Lancet 2003;362:383-91.) |
CLINICAL PRESENTATION
The classic clinical presentation of celiac disease consists of diarrhea, gas and bloating, and weight loss (30). Since the 1980s, investigators have noted a shift in presentation of celiac disease in the United States from diarrhea to asymptomatic patients detected at screening (31) or as iron deficiency anemia or bone disease. Other presentations
include fatigue, constipation, dyspepsia, unexplained hepatitis, neuropathy, ataxia, dental hypoplasia, and infertility. A 50-fold higher risk of microscopic colitis exists in celiac disease (32). Virtually all individuals with the burning, itching rash of dermatitis herpetiformis have celiac disease (33). Individuals with celiac disease may be obese on diagnosis (34). High-risk groups for the development of celiac disease include those with affected first-degree relatives, type 1 diabetes mellitus, autoimmune thyroid disease, irritable bowel syndrome, primary biliary cirrhosis, multiple sclerosis, and Down, William, and Turner syndromes (35). Most individuals with asymptomatic celiac disease have been identified by screening first-degree relatives of patients with celiac disease.
include fatigue, constipation, dyspepsia, unexplained hepatitis, neuropathy, ataxia, dental hypoplasia, and infertility. A 50-fold higher risk of microscopic colitis exists in celiac disease (32). Virtually all individuals with the burning, itching rash of dermatitis herpetiformis have celiac disease (33). Individuals with celiac disease may be obese on diagnosis (34). High-risk groups for the development of celiac disease include those with affected first-degree relatives, type 1 diabetes mellitus, autoimmune thyroid disease, irritable bowel syndrome, primary biliary cirrhosis, multiple sclerosis, and Down, William, and Turner syndromes (35). Most individuals with asymptomatic celiac disease have been identified by screening first-degree relatives of patients with celiac disease.
DIAGNOSIS
Small Intestinal Biopsy
Small intestinal biopsy remains the gold standard in the diagnosis of celiac disease. Varying degrees of inflammation may be present. In the mildest form, villous architecture is normal, and an increase in intraepithelial lymphocytes is noted at the villous tips. Most individuals with celiac disease have villous atrophy with crypt hyperplasia as well as increased intraepithelial lymphocytes and increased lymphocytes and plasma cells in the lamina propria (7, 33). These inflammatory changes are not specific for celiac disease, especially the mildest forms; therefore, diagnosis is based on antibody testing and histologic improvement on a gluten-free diet (7). Celiac disease mainly affects the proximal small bowel; the first site of gluten exposure is the duodenum. Multiple duodenal biopsies during a glutencontaining diet are recommended to ensure adequate tissue for diagnosis (35). The correlation between duodenal histologic features and clinical symptoms is poor (36), perhaps because of the length of the intestine and the degree of inflammatory involvement.
Antibody Testing
Over the years, certain antibodies have been identified that are associated with celiac disease, most are of the immunoglobulin (IgA) class (37). These antibodies are useful to support the diagnosis of celiac disease, screen high-risk groups, and monitor for adherence to a gluten-free diet, particularly in individuals with ongoing symptoms. The endomysial antibody (EMA) and tTG IgA antibody have the highest sensitivity and specificity for celiac disease in adults and children (5). tTG is the autoantigen recognized by EMA (37, 38). Deamidated gliadin peptide (DGP) IgA and IgG antibody tests that recognize recombinant toxic gliadin peptides deamidated by tTG have been developed. These tests have a sensitivity and specificity approaching those of EMA and tTG antibodies. The DGP IgG antibody test is particularly useful in diagnosing celiac disease in persons with complete IgA deficiency because it has a higher sensitivity and specificity than tTG IgG antibody (37). The tTG IgA antibody has a lower positive predictive value for celiac disease than does EMA because of falsepositive test results found in other inflammatory conditions such as Crohn disease, ulcerative colitis, and primary biliary cirrhosis (39, 40).
The antigliadin IgA antibody (AGA) has poor sensitivity and specificity for celiac disease and is no longer used (37). A positive AGA test result may be useful as a marker of gluten sensitivity or intolerance (41), a label given to individuals with gastrointestinal symptoms, negative tTG and EMA antibodies, and a normal duodenal biopsy who improve on a gluten-free diet.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


