29
Catabolism of the Carbon Skeletons of Amino Acids
OBJECTIVES
After studying this chapter, you should be able to:
![]() Name the principal catabolites of the carbon skeletons of the common amino acids and the major metabolic fates of these catabolites.
Name the principal catabolites of the carbon skeletons of the common amino acids and the major metabolic fates of these catabolites.
![]() Write an equation for an aminotransferase (transaminase) reaction and illustrate the role played by the coenzyme.
Write an equation for an aminotransferase (transaminase) reaction and illustrate the role played by the coenzyme.
![]() Outline the metabolic pathways for each of the common amino acids, and identify reactions associated with clinically significant metabolic disorders.
Outline the metabolic pathways for each of the common amino acids, and identify reactions associated with clinically significant metabolic disorders.
![]() Provide examples of aminoacidurias that arise from defects in glomerular tubular reabsorption, and the consequences of impaired intestinal absorption of tryptophan.
Provide examples of aminoacidurias that arise from defects in glomerular tubular reabsorption, and the consequences of impaired intestinal absorption of tryptophan.
![]() Explain why metabolic defects in different enzymes of the catabolism of a specific amino acid can be associated with similar clinical signs and symptoms.
Explain why metabolic defects in different enzymes of the catabolism of a specific amino acid can be associated with similar clinical signs and symptoms.
![]() Describe the implications of a metabolic defect in glutamate-γ-semialdehyde dehydrogenase for the catabolism of proline and of 4-hydroxyproline.
Describe the implications of a metabolic defect in glutamate-γ-semialdehyde dehydrogenase for the catabolism of proline and of 4-hydroxyproline.
![]() Explain how the α-amino nitrogen of proline and of lysine is removed by processes other than transamination.
Explain how the α-amino nitrogen of proline and of lysine is removed by processes other than transamination.
![]() Draw analogies between the reactions that participate in the catabolism of fatty acids and of the branched-chain amino acids.
Draw analogies between the reactions that participate in the catabolism of fatty acids and of the branched-chain amino acids.
![]() Identify the specific metabolic defects in hypervalinemia, maple syrup urine disease, intermittent branched-chain ketonuria, isovaleric acidemia, and methylmalonic aciduria.
Identify the specific metabolic defects in hypervalinemia, maple syrup urine disease, intermittent branched-chain ketonuria, isovaleric acidemia, and methylmalonic aciduria.
BIOMEDICAL IMPORTANCE
The prior chapter described the removal and metabolic fate of the nitrogen atoms of the common L-α-amino acids. This chapter will address the metabolic fates of the resulting hydrocarbon skeletons of these amino acids. Discussed are the enzymes and intermediates formed during the conversion of the carbon skeletons to amphibolic intermediates, and several metabolic diseases or “inborn errors of metabolism” associated with these processes. While most disorders of amino acid catabolism are rare, if left untreated they can result in irreversible brain damage and early mortality. Prenatal or early postnatal detection of metabolic disorders and timely initiation of treatment thus are essential. The ability to detect the activities of enzymes in cultured amniotic fluid cells facilitates prenatal diagnosis by amniocentesis. All states now conduct screening tests of newborns for as many as 30 metabolic diseases. These tests include, but are not limited to, disorders associated with defects in the catabolism of amino acids. The most reliable screening tests use tandem mass spectrometry to detect, in a few drops of neonate blood, catabolites suggestive of a given metabolic defect. The metabolites detected pinpoint the metabolic defect as the lowered or absent activity of a given enzyme. Treatment consists primarily of feeding diets low in the amino acid whose catabolism is impaired.
Mutations in the exons or in the regulatory regions of a gene that encodes an enzyme of amino acid metabolism can result in the failure to synthesize that enzyme or in the synthesis of a partially or completely nonfunctional enzyme. Mutations may have no significant effect of the activity of the encoded enzyme. By contrast, mutations that compromise the overall three-dimensional structure or the structure of catalytic or regulatory sites may be associated with adverse metabolic consequences. Low catalytic efficiency of a mutant enzyme can result from impaired positioning of residues involved in catalysis, or in binding a substrate, coenzyme, or metal ion. Mutations may also impair the ability of certain enzymes to respond appropriately to the signals that modulate their activity by altering an enzyme’s affinity for an allosteric regulator of activity. Since different mutations can have similar effects on any of the above factors, various mutations may give rise to the same clinical signs and symptoms. At a molecular level, these therefore are distinct molecular diseases. To supplement the disorders of amino acid metabolism discussed in this chapter, readers should consult major reference works on this topic such as Scriver et al 2001.
AMINO ACIDS ARE CATABOLIZED TO INTERMEDIATES FOR CARBOHYDRATE AND LIPID BIOSYNTHESIS
Nutritional studies in the period 1920-1940, reinforced and confirmed by studies using isotopically labeled amino acids conducted from 1940 to 1950, established the interconvertibility of the carbon atoms of fat, carbohydrate, and protein. These studies also revealed that all or a portion of the carbon skeleton of every amino acid is convertible either to carbohydrate (13 amino acids), fat (one amino acid), or both fat and carbohydrate (five amino acids) (Table 29-1). Figure 29–1 outlines overall aspects of these interconversions.
TABLE 29–1 Fate of the Carbon Skeletons of the Common L-α-Amino Acids
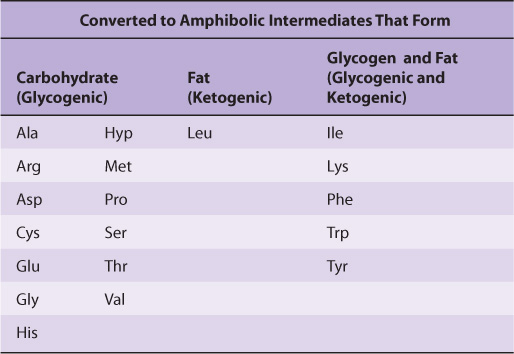
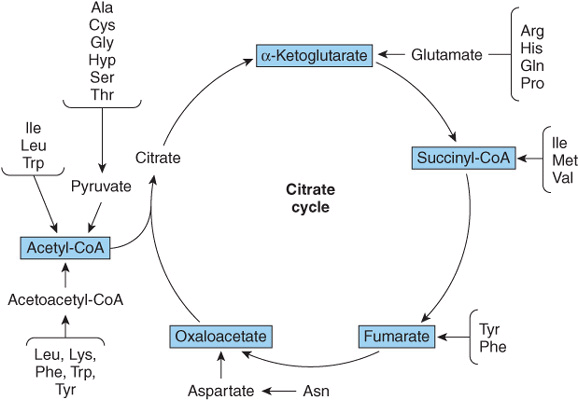
FIGURE 29–1 Overview of the amphibolic intermediates that result from catabolism of the common amino acids.
TRANSAMINATION TYPICALLY INITIATES AMINO ACID CATABOLISM
Removal of α-amino nitrogen by transamination, a reaction catalyzed by an aminotransferase or transaminase (see Figure 28–6), is the first catabolic reaction of all the common amino acids except proline, hydroxyproline, threonine, or lysine. The hydrocarbon skeleton that remains is then degraded to amphibolic intermediates as outlined in Figure 29–1.
Asparagine and Aspartate Form Oxaloacetate
All four carbons of asparagine and of aspartate form oxaloacetate via reactions catalyzed by asparaginase and a transaminase (Figure 29–2, top). Metabolic defects in transaminases, which fulfill central amphibolic functions, may be incompatible with life. Consequently, no known metabolic defect is associated with this short catabolic pathway.
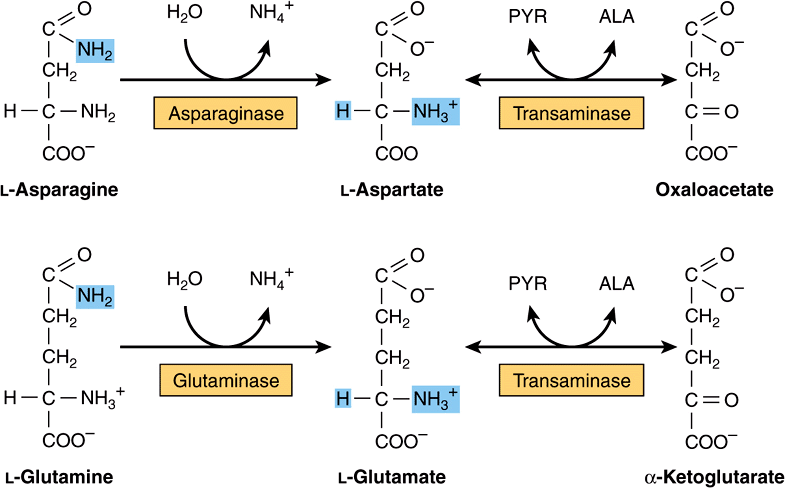
FIGURE 29–2 Catabolism to amphibolic intermediates of L-asparagine (top) and of L-glutamine (bottom). (PYR, pyruvate; ALA, L-alanine.) In this and subsequent figures, blue highlights emphasize the portions of the molecules that are undergoing chemical change.
Glutamine and Glutamate Form α-Ketoglutarate
The catabolism of glutamine and of glutamate parallels that of asparagine and aspartate in reactions catalyzed by glutaminase and transaminase that forms α-ketoglutarate (Figure 29–2, bottom). While both glutamate and aspartate are substrates for the same transaminase, deamidation of their corresponding amides is catalyzed by different enzymes: asparaginase and glutaminase. Possibly for the reason stated earlier, there are no known metabolic defects of the glutamine-glutamate catabolic pathway.
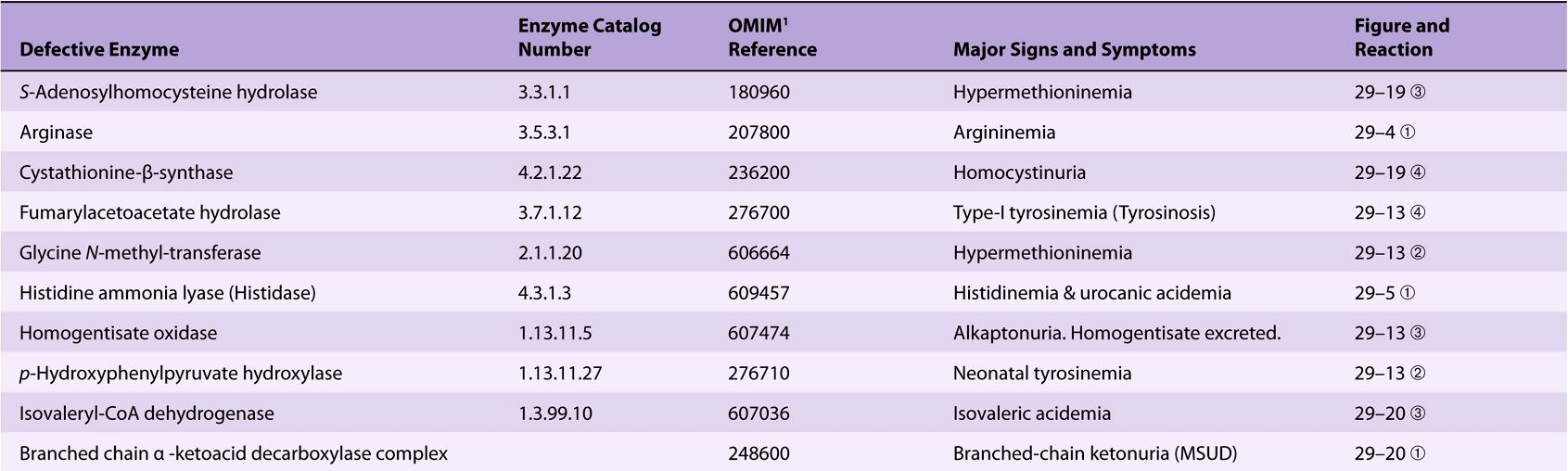
Significant metabolic disorders are, however, associated with the catabolism of many other amino acids. Discussed below under the catabolism of each amino acid, these disorders are summarized in Table 29-2. This table lists the impaired enzyme, its IUB enzyme catalog (EC) number, a cross-reference to a specific figure and numbered reaction, and a numerical link to the Online Mendelian Inheritance in Man database (OMIM).
TABLE 29–2 Metabolic Diseases of Amino Acid Metabolism
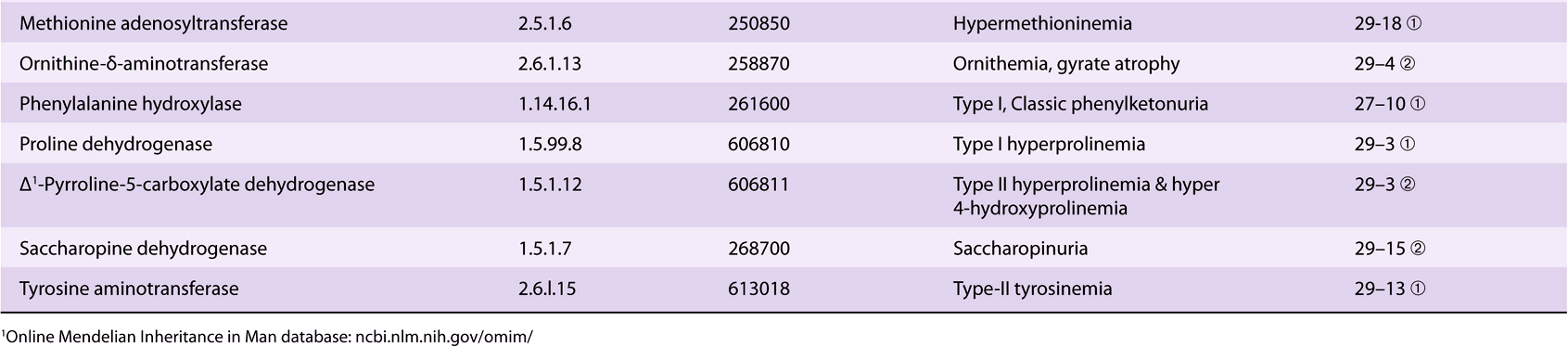
Proline
The catabolism of proline takes place in mitochondria. Since proline does not participate in transamination, the nitrogen of this imino acid is retained throughout its oxidation to Δ1-pyrrolline-5-carboxylate, ring opening to glutamate-γ-semialdehyde, and oxidation to glutamate, and is only removed during transamination of glutamate to a-ketoglutarate (Figure 29–3). There are two metabolic disorders of proline catabolism. Both are inherited as autosomal recessive traits and are consistent with a normal adult life. The metabolic block in type I hyperprolinemia is at proline dehydrogenase. There is no associated impairment of hydroxyproline catabolism. The metabolic block in type II hyperprolinemia is at glutamate-γ-semialdehyde dehydrogenase, a mitochondrial matrix enzyme that also participates in the catabolism of arginine, ornithine, and hydroxyproline (see below). Since proline and hydroxyproline catabolism are affected, both Δ1-pyrroline-5-carboxylate and Δ1-pyrroline-3-hydroxy-5-carboxylate (see Figure 29–12) are excreted.
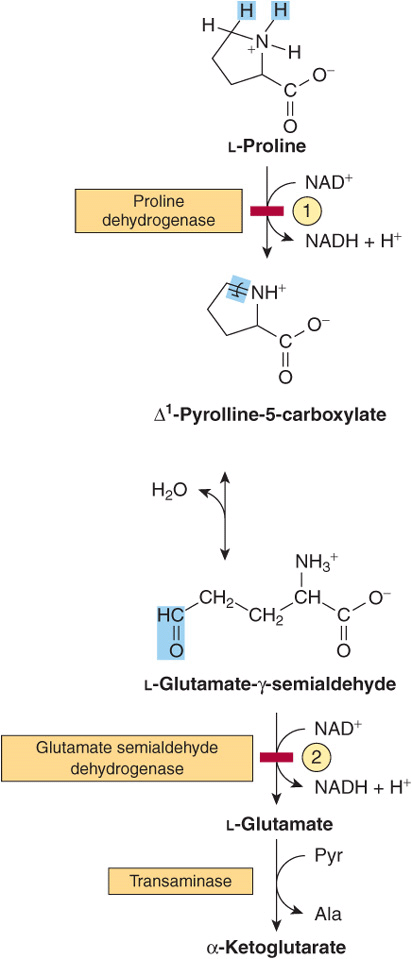
FIGURE 29–3 Catabolism of proline. Red bars and circled numerals indicate the locus of the inherited metabolic defects in ![]() type-I hyperprolinemia and
type-I hyperprolinemia and ![]() type-II hyperprolinemia.
type-II hyperprolinemia.
Arginine and Ornithine
The initial reactions in arginine catabolism are conversion to ornithine followed by transamination of ornithine to glutamate-γ-semialdehyde (Figure 29–4). Subsequent catabolism of glutamate-γ-semialdehyde to α-ketoglutarate occurs as described for proline (see Figure 29–3). Mutations in ornithine δ-aminotransferase (ornithine transaminase) elevate plasma and urinary ornithine and are associated with gyrate atrophy of the choroid and retina. Treatment involves restricting dietary arginine. In the hyperornithinemia-hyperammonemia syndrome, a defective mitochondrial ornithine-citrulline antiporter (see Figure 28–13) impairs transport of ornithine into mitochondria for use in urea synthesis.
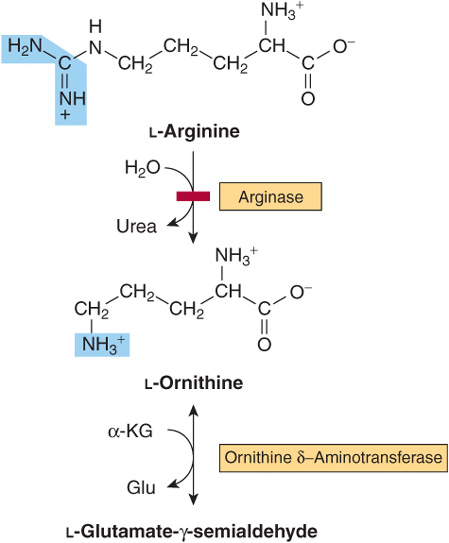
FIGURE 29–4 Catabolism of arginine. Arginase-catalyzed cleavage of L-arginine forms urea and L-ornithine. This reaction (red bar) represents the site of the inherited metabolic defect in hyperargininemia. Subsequent transamination of L-ornithine to glutamate-γ-semialdehyde is followed by conversion to α-ketoglutarate (see Figure 29–3).
Histidine
Catabolism of histidine proceeds via urocanate, 4-imida-zolone-5-propionate, and N-formiminoglutamate (Figlu). Formimino group transfer to tetrahydrofolate forms glutamate, then α-ketoglutarate (Figure 29–5). In folic acid deficiency, transfer of the formimino group is impaired, and Figlu is excreted. Excretion of Figlu following a dose of histidine thus can be used to detect folic acid deficiency. Benign disorders of histidine catabolism include histidinemia and urocanic aciduria associated with impaired histidase.
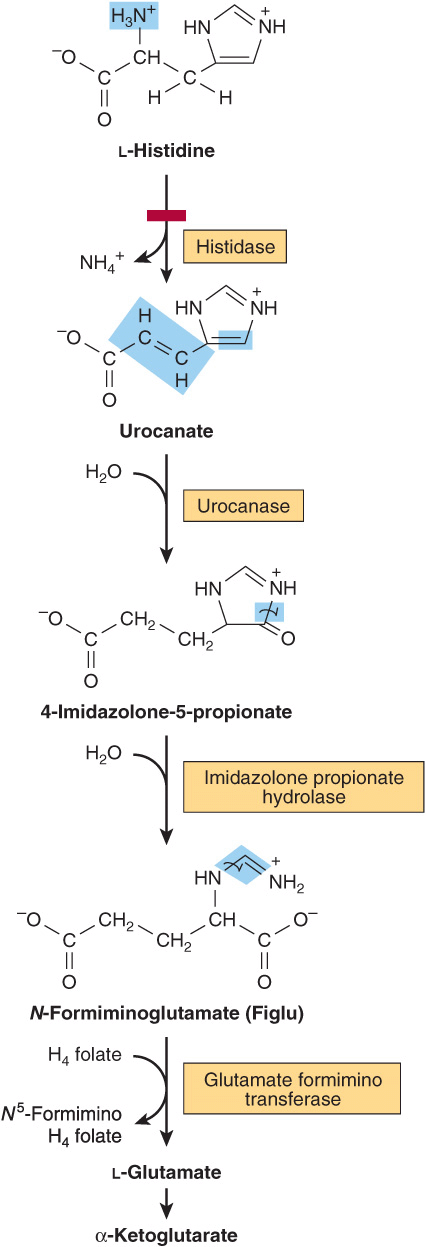
FIGURE 29–5 Catabolism of L-histidine to α-ketoglutarate. (H4 folate, tetrahydrofolate.) The red bar indicates the site of an inherited metabolic defect.
CATABOLISM OF GLYCINE, SERINE, ALANINE, CYSTEINE, THREONINE, AND 4HYDROXYPROLINE
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


