KEY CONCEPTS
![]() Carcinogenesis is a multistep process that includes initiation, promotion, conversion, and progression. The growth of normal and cancerous cells is genetically controlled by the balance or imbalance of oncogene and tumor suppressor gene protein products. Multiple genetic mutations are required to convert normal cells to cancerous cells. Apoptosis and cellular senescence (aging) are normal mechanisms for cell death.
Carcinogenesis is a multistep process that includes initiation, promotion, conversion, and progression. The growth of normal and cancerous cells is genetically controlled by the balance or imbalance of oncogene and tumor suppressor gene protein products. Multiple genetic mutations are required to convert normal cells to cancerous cells. Apoptosis and cellular senescence (aging) are normal mechanisms for cell death.
![]() Several signaling pathways are dysregulated in many common cancers. Several agents have been developed to prevent signal transduction through these pathways. Monoclonal antibodies, which competitively bind to extracellular receptors or their natural ligands, and targeted drugs, which target a component of the intracellular signal transduction pathway, are available for several cancers.
Several signaling pathways are dysregulated in many common cancers. Several agents have been developed to prevent signal transduction through these pathways. Monoclonal antibodies, which competitively bind to extracellular receptors or their natural ligands, and targeted drugs, which target a component of the intracellular signal transduction pathway, are available for several cancers.
![]() Tumors must develop new blood vessels through the process of angiogenesis in order to grow. This process, regulated by proangiogenic and antiangiogenic factors, becomes dysregulated in several cancers and can lead to tumor growth, invasion, and metastasis. New anticancer agents can target this process and decrease tumor growth.
Tumors must develop new blood vessels through the process of angiogenesis in order to grow. This process, regulated by proangiogenic and antiangiogenic factors, becomes dysregulated in several cancers and can lead to tumor growth, invasion, and metastasis. New anticancer agents can target this process and decrease tumor growth.
![]() Because patients with clinically evident metastatic cancer can rarely be cured, early detection is critical. Screening programs are designed to detect cancers in asymptomatic people who are at risk of a specific cancer. Knowing the early warning signs of cancer is also important in early detection, when cancers are most likely to be localized.
Because patients with clinically evident metastatic cancer can rarely be cured, early detection is critical. Screening programs are designed to detect cancers in asymptomatic people who are at risk of a specific cancer. Knowing the early warning signs of cancer is also important in early detection, when cancers are most likely to be localized.
![]() Treatment for cancer should not begin until the presence of cancer is confirmed by a tissue (e.g., histologic) diagnosis. Clinical cancer staging provides prognostic information, and in conjunction with the patient’s treatment goals, guides the selection of anticancer treatment. The goals include cure, prolongation of life, and palliation. Surgery and radiation provide the best chance of cure for patients with localized cancers, but systemic treatment methods are required for disseminated cancers.
Treatment for cancer should not begin until the presence of cancer is confirmed by a tissue (e.g., histologic) diagnosis. Clinical cancer staging provides prognostic information, and in conjunction with the patient’s treatment goals, guides the selection of anticancer treatment. The goals include cure, prolongation of life, and palliation. Surgery and radiation provide the best chance of cure for patients with localized cancers, but systemic treatment methods are required for disseminated cancers.
![]() Adjuvant therapy is systemic therapy that is administered to treat any existing micrometastases remaining after surgical excision of localized disease. Because adjuvant therapy is given to patients with no remaining clinical evidence of cancer, the benefit of the treatment cannot be proven for an individual patient but only for patient populations. Treatment decisions are based largely on an assessment of the presence of risk factors in an individual patient and their estimated risk for cancer recurrence. The effectiveness of adjuvant therapy is measured by the relative and absolute reduction in the risk of recurrence.
Adjuvant therapy is systemic therapy that is administered to treat any existing micrometastases remaining after surgical excision of localized disease. Because adjuvant therapy is given to patients with no remaining clinical evidence of cancer, the benefit of the treatment cannot be proven for an individual patient but only for patient populations. Treatment decisions are based largely on an assessment of the presence of risk factors in an individual patient and their estimated risk for cancer recurrence. The effectiveness of adjuvant therapy is measured by the relative and absolute reduction in the risk of recurrence.
![]() Traditional chemotherapy affects rapidly proliferating cells. Chemotherapy can be either “cell-cycle phase specific,” targeting one specific phase of the cell cycle, or “cell-cycle phase nonspecific,” targeting all proliferating cells regardless of their place in the cell cycle. Whereas cell-cycle phase-specific chemotherapies are generally given more frequently or as continuous infusions, cell-cycle phase-nonspecific chemotherapies are usually given as a single dose.
Traditional chemotherapy affects rapidly proliferating cells. Chemotherapy can be either “cell-cycle phase specific,” targeting one specific phase of the cell cycle, or “cell-cycle phase nonspecific,” targeting all proliferating cells regardless of their place in the cell cycle. Whereas cell-cycle phase-specific chemotherapies are generally given more frequently or as continuous infusions, cell-cycle phase-nonspecific chemotherapies are usually given as a single dose.
![]() Monoclonal antibodies recognize an antigen that is expressed preferentially on cancer cells or target growth factors responsible for cancer growth. These therapies can vary in the amount of foreign component that can be used to predict tolerability. Monoclonal antibodies that target cellular antigens induce cell death by a variety of mechanisms that involve the host immune system. These antibodies can also be used to deliver drugs, radioisotopes, or toxins to the antigen-expressing cells.
Monoclonal antibodies recognize an antigen that is expressed preferentially on cancer cells or target growth factors responsible for cancer growth. These therapies can vary in the amount of foreign component that can be used to predict tolerability. Monoclonal antibodies that target cellular antigens induce cell death by a variety of mechanisms that involve the host immune system. These antibodies can also be used to deliver drugs, radioisotopes, or toxins to the antigen-expressing cells.
![]() Understanding the mechanism of toxicities can lead to more effective prevention and treatment of these toxicities. Prospective dose modification of some chemotherapy and targeted therapies are essential in patients with impaired organ function to reduce the risk of severe adverse events. Identification of genetic variations that affect activation and metabolism may permit the development of individualized therapy that optimize effectiveness and minimize toxicity.
Understanding the mechanism of toxicities can lead to more effective prevention and treatment of these toxicities. Prospective dose modification of some chemotherapy and targeted therapies are essential in patients with impaired organ function to reduce the risk of severe adverse events. Identification of genetic variations that affect activation and metabolism may permit the development of individualized therapy that optimize effectiveness and minimize toxicity.
![]() Myelosuppression is the acute dose-limiting toxicity for most nonspecific chemotherapy. Whereas anemia can cause fatigue in patients with cancer, the risk of infection in patients is related to the depth and duration of neutropenia. Unexplained fever in neutropenic patients requires prompt initiation of empiric antibiotic therapy. Colony-stimulating factors are available to improve fatigue in patients with anemia and reduce the risk of febrile neutropenia. Evidence-based clinical guidelines should direct the use of these supportive care measures.
Myelosuppression is the acute dose-limiting toxicity for most nonspecific chemotherapy. Whereas anemia can cause fatigue in patients with cancer, the risk of infection in patients is related to the depth and duration of neutropenia. Unexplained fever in neutropenic patients requires prompt initiation of empiric antibiotic therapy. Colony-stimulating factors are available to improve fatigue in patients with anemia and reduce the risk of febrile neutropenia. Evidence-based clinical guidelines should direct the use of these supportive care measures.
INTRODUCTION
Cancer is a group of more than 100 different diseases that are characterized by uncontrolled cellular growth, local tissue invasion, and distant metastases.1 It is now the leading cause of death in Americans younger than age 85 years. Nearly 1.7 million cases of cancer were projected for 2013 with an estimated 580,350 lives claimed in the United States.2 Figure 104-1 illustrates the estimated incidence of common cancers and cancer-related deaths. The four most common cancers are prostate, breast, lung, and colorectal cancer. The most common cause of cancer-related deaths in the United States is lung cancer, which accounts for about 160,000 deaths each year. These cancers are discussed in further detail in the chapters that follow.
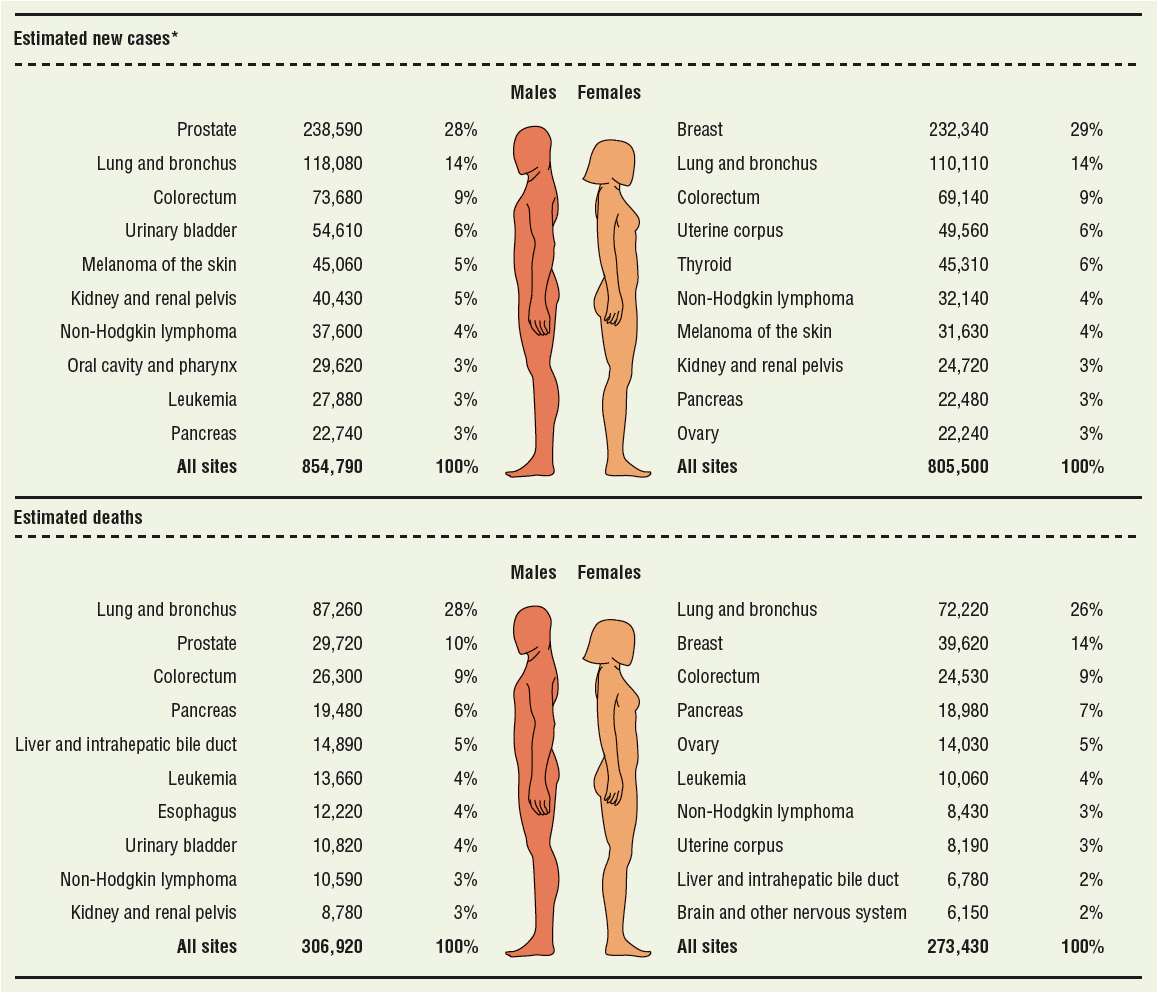
FIGURE 104-1 Estimated 2013 cancer incidences (top) and deaths (bottom) in the United States for males and females. *Estimate are rounded to the nearest 10 and exclude basal cell and squamous cell skin cancers and in situ carcinoma except urinary bladder. (Reproduced with permission from Siegel et al.2)
The roles of healthcare providers in the management of patients with cancer can be very diverse. Thorough knowledge of the pharmacology and the pharmacokinetics of anticancer agents is essential to prevent and manage toxicities. Supportive care issues, such as nutritional support, pain management, infection, and nausea and vomiting, require application of clinical, pharmacologic, and economic principles. Provision of drug information to other healthcare providers and to patients and their families is another critical role. Experienced healthcare providers are able to fulfill these roles and make valuable contributions to patient care in the oncology setting.
This chapter introduces the basic concepts of carcinogenesis, tumor growth, and anticancer treatment; provides general information on the pharmacology and clinical use of anticancer agents; and presents an overview of supportive care issues.
ETIOLOGY OF CANCER
Carcinogenesis
![]() The mechanisms by which cancers occur are incompletely understood. A cancer is thought to develop from a cell in which the normal mechanisms for control of growth and proliferation are altered. Current evidence supports the concept of carcinogenesis as a multistage process that is genetically regulated.3–6 The first step in this process is initiation, which requires exposure of normal cells to carcinogenic substances. These carcinogens produce genetic damage that, if not repaired, results in irreversible cellular mutations. This mutated cell has an altered response to its environment and a selective growth advantage, giving it the potential to develop into a clonal population of neoplastic cells. During the second phase, known as promotion, carcinogens or other factors alter the environment to favor growth of the mutated cell population over normal cells. The primary difference between initiation and promotion is that promotion is a reversible process. Because it is reversible, the promotion phase may be the target of future chemoprevention strategies, including changes in lifestyle and diet. At some point, however, the mutated cell becomes cancerous (conversion or transformation). Depending on the cancer, 5 to 20 years may elapse between the initiation and promotion and the development of a clinically detectable cancer. The final stage of neoplastic growth, called progression, involves further genetic changes leading to increased cell proliferation. The critical elements of this phase include tumor invasion into local tissues and the development of metastases.
The mechanisms by which cancers occur are incompletely understood. A cancer is thought to develop from a cell in which the normal mechanisms for control of growth and proliferation are altered. Current evidence supports the concept of carcinogenesis as a multistage process that is genetically regulated.3–6 The first step in this process is initiation, which requires exposure of normal cells to carcinogenic substances. These carcinogens produce genetic damage that, if not repaired, results in irreversible cellular mutations. This mutated cell has an altered response to its environment and a selective growth advantage, giving it the potential to develop into a clonal population of neoplastic cells. During the second phase, known as promotion, carcinogens or other factors alter the environment to favor growth of the mutated cell population over normal cells. The primary difference between initiation and promotion is that promotion is a reversible process. Because it is reversible, the promotion phase may be the target of future chemoprevention strategies, including changes in lifestyle and diet. At some point, however, the mutated cell becomes cancerous (conversion or transformation). Depending on the cancer, 5 to 20 years may elapse between the initiation and promotion and the development of a clinically detectable cancer. The final stage of neoplastic growth, called progression, involves further genetic changes leading to increased cell proliferation. The critical elements of this phase include tumor invasion into local tissues and the development of metastases.
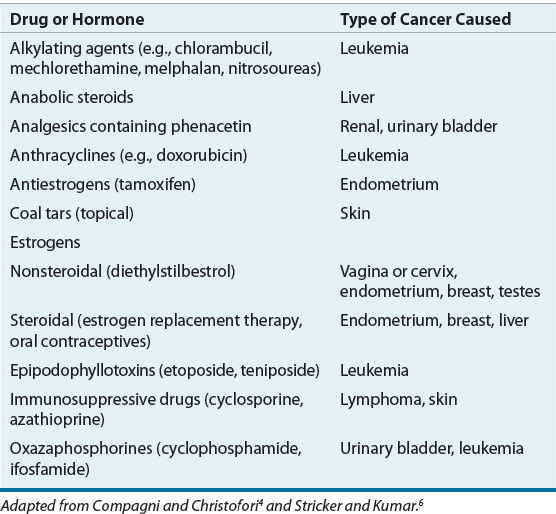
Substances that may act as carcinogens or initiators include chemical, physical, and biologic agents.5 Exposure to chemicals may occur by virtue of occupational and environmental means, as well as lifestyle habits. The association of aniline dye exposure and bladder cancer is one such example. Benzene is known to cause leukemia. Some drugs and hormones used for therapeutic purposes are also classified as carcinogenic chemicals (Table 104-1). Physical agents that act as carcinogens include ionizing radiation and ultraviolet light; radiation induces mutations by forming free radicals that damage DNA (deoxyribonucleic acid) and other cellular components. Viruses are biologic agents that are associated with certain cancers. The Epstein-Barr virus (EBV) is believed to be an important factor in the initiation of Burkitt lymphoma. Likewise, infection with human papilloma virus (HPV) is known to be a major cause of cervical cancer and head and neck cancers. All of the previously mentioned carcinogens, as well as age, gender, diet, growth factors, and chronic irritation, are among the factors considered to be promoters of carcinogenesis.
TABLE 104-1 Selected Drugs and Hormones Known to Cause Cancer in Humans
Genetic and Molecular Basis of Cancer
![]() In recent years, there has been marked progress in our understanding of the genetic changes that lead to the development of cancer, largely because of improvements in research techniques and new genomic information.3,5–7 Two major classes of genes are involved in carcinogenesis: oncogenes and tumor suppressor genes. Figure 104-2 illustrates the acquired capabilities of cancer cells that differ from normal cellular function.8 Oncogenes develop from normal genes, called protooncogenes, and may have important roles in all phases of carcinogenesis. Protooncogenes are present in all cells and are essential regulators of normal cellular functions, including the cell cycle. Genetic alteration of the protooncogene through point mutation, chromosomal rearrangement, or gene amplification activates the oncogene. These genetic alterations may be caused by carcinogenic agents such as radiation, chemicals, or viruses (somatic mutations), or they may be inherited (germ-line mutations). After activation, the oncogene produces either excessive amounts of the normal gene product or an abnormal gene product. The result is dysregulation of normal cell growth and proliferation, which imparts a distinct growth advantage to the cell and increases the probability of neoplastic transformation. An example is the human epidermal growth factor receptor (HER) family of oncogenes. This family of receptor tyrosine kinases contains four members: epidermal growth factor receptor (EGFR), HER2, HER3, and HER4. When activated, these receptors mediate cell proliferation and differentiation of cells through activation of intracellular tyrosine kinase receptors and downstream signaling pathways. As an oncogene, the gene product is overexpressed or amplified, resulting in excessive cellular proliferation, metastasis, angiogenesis, and cell survival in several cancers. Table 104-2 lists examples of oncogenes by their cellular function.9
In recent years, there has been marked progress in our understanding of the genetic changes that lead to the development of cancer, largely because of improvements in research techniques and new genomic information.3,5–7 Two major classes of genes are involved in carcinogenesis: oncogenes and tumor suppressor genes. Figure 104-2 illustrates the acquired capabilities of cancer cells that differ from normal cellular function.8 Oncogenes develop from normal genes, called protooncogenes, and may have important roles in all phases of carcinogenesis. Protooncogenes are present in all cells and are essential regulators of normal cellular functions, including the cell cycle. Genetic alteration of the protooncogene through point mutation, chromosomal rearrangement, or gene amplification activates the oncogene. These genetic alterations may be caused by carcinogenic agents such as radiation, chemicals, or viruses (somatic mutations), or they may be inherited (germ-line mutations). After activation, the oncogene produces either excessive amounts of the normal gene product or an abnormal gene product. The result is dysregulation of normal cell growth and proliferation, which imparts a distinct growth advantage to the cell and increases the probability of neoplastic transformation. An example is the human epidermal growth factor receptor (HER) family of oncogenes. This family of receptor tyrosine kinases contains four members: epidermal growth factor receptor (EGFR), HER2, HER3, and HER4. When activated, these receptors mediate cell proliferation and differentiation of cells through activation of intracellular tyrosine kinase receptors and downstream signaling pathways. As an oncogene, the gene product is overexpressed or amplified, resulting in excessive cellular proliferation, metastasis, angiogenesis, and cell survival in several cancers. Table 104-2 lists examples of oncogenes by their cellular function.9
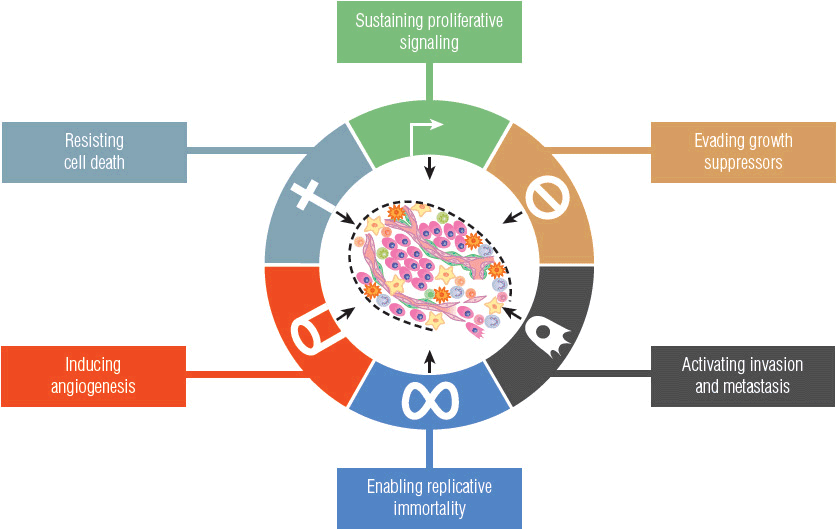
FIGURE 104-2 Functional capabilities acquired by cancer cells, including angiogenesis, self-proliferation, insensitivity to antigrowth signals and limitless growth potential, metastasis, and antiapoptotic effects. It is thought that most, if not all, cancer cells acquire these functions through a variety of mechanisms, including activation of oncogenes and mutations in tumor suppressor genes. (Reprinted from Cell, Vol 144(5), Hanahan D, Weinberg RA, The Hallmarks of Cancer: The Next Generation, Copyright © 2011, with permission from Elsevier.)
TABLE 104-2 Examples of Oncogenes and Tumor Suppressor Genes
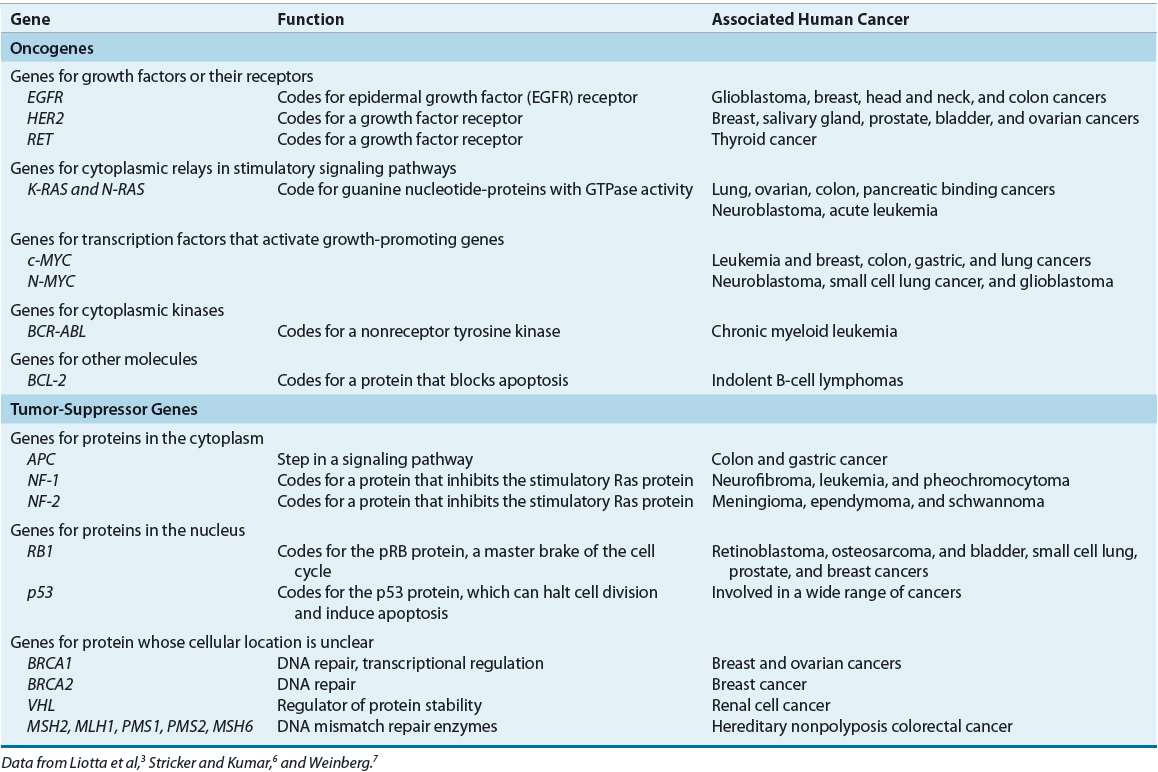
In contrast, tumor suppressor genes regulate and inhibit inappropriate cellular growth and proliferation.3,6,7 Gene loss or mutation results in loss of control over normal cell growth. Two common examples of tumor suppressor genes are the retinoblastoma (Rb) and p53 genes. Mutation of p53 is one of the most common genetic changes associated with cancer, and it is estimated to occur in half of all cancers.7 The normal gene product of p53 is responsible for negative regulation of the cell cycle, allowing the cell cycle to halt for repairs, corrections, and responses to other external signals. Inactivation of p53 removes this checkpoint, allowing mutations to occur. Mutation of p53 is linked to a variety of cancers, including brain tumors (astrocytoma); carcinomas of the breast, colon, lung, cervix, and anus; and osteosarcoma. Another important function of p53 may be modulation of cytotoxic drug effects. Loss of p53 is associated with anticancer drug resistance.
Another group of genes important in carcinogenesis are the DNA repair genes. The normal function of these genes is to repair DNA that is damaged by environmental factors or errors in DNA that occur during replication.6 If not corrected, these errors can result in mutations that activate oncogenes or inactivate tumor suppressor genes. As more mutations in the genome occur, the risk for malignant transformation increases. The DNA repair genes have been classified as tumor suppressor genes, because a loss in their function results in an increased risk for carcinogenesis. Deficiencies in DNA repair genes have been discovered in familial colon cancer (hereditary nonpolyposis colon cancer) and breast cancer.
![]() Oncogenes and tumor suppressor genes provide the stimulatory and inhibitory signals that ultimately regulate the cell cycle.3,7 These signals converge on a molecular system in the nucleus known as the cell-cycle clock. The function of the clock in normal tissue is to integrate the signal input and to determine if the cell cycle should proceed. The clock is composed of a series of interacting proteins, the most important of which are cyclins and cyclin-dependent kinases (CDKs). Cyclins (especially cyclin D1) and CDKs promote entry into the cell cycle and are overexpressed in several cancers, including breast cancer. CDK inhibitors have been identified as important negative regulators of the cell cycle.
Oncogenes and tumor suppressor genes provide the stimulatory and inhibitory signals that ultimately regulate the cell cycle.3,7 These signals converge on a molecular system in the nucleus known as the cell-cycle clock. The function of the clock in normal tissue is to integrate the signal input and to determine if the cell cycle should proceed. The clock is composed of a series of interacting proteins, the most important of which are cyclins and cyclin-dependent kinases (CDKs). Cyclins (especially cyclin D1) and CDKs promote entry into the cell cycle and are overexpressed in several cancers, including breast cancer. CDK inhibitors have been identified as important negative regulators of the cell cycle.
The cell cycle proceeds from one cell division to the next. The cycle involves five phases: DNA replication (S phase), cell division (M phase), two resting phases (G1, G2), and a nondividing state (G0 phase). In the first resting phase G1, the cell grows in size and decides to commit to the cell cycle or remain in a resting state. If the cell is normal the cell will move into the S phase to synthesize its DNA. Next, the cell enters the second resting phase G2, in which the cell prepares to divide. In the M phase, the cell enters mitosis and yields two daughter cells. If the cell is not normal the cell can stop dividing and initiate apoptosis.
Four checkpoints exist within the cell cycle, one in each phase of the cell cycle, and serve as quality control checkpoints. The cell will not proceed to the next phase unless all requirements for the current phase are met. Complexes of cyclin and CDK regulate these checkpoints. These complexes lead to the activation of other proteins that are responsible for the specific events of each phase of the cell cycle. The first checkpoint is called the restriction site. The restriction site is controlled by Rb complexed to a transcription factor called E2F. The presence of this complex prevents cell-cycle progression. A cell can proceed beyond the G1 restriction site and continue into the S phase, when cyclin–CDK complexes phosphorylate Rb and target it for degradation. A cell may alternatively withdraw into the G0 phase in the presence of antimitogenic or the absence of mitogenic factors.10
![]() When the normal regulatory mechanisms for cellular growth fail, backup defense systems may be activated. The secondary defenses include apoptosis (programmed cell death or suicide) and cellular senescence (aging). Apoptosis is a normal mechanism of cell death required for tissue homeostasis.3,7,11 This process is regulated by oncogenes and tumor suppressor genes and is also a mechanism of cellular death after exposure to cytotoxins. Overexpression of oncogenes responsible for apoptosis may produce an “immortal” cell, which has increased potential for malignancy. The bcl-2 oncogene is an example. The most common chromosomal abnormality found in lymphoid malignancies is the t(14;18) translocation. The bcl-2 protooncogene is normally located on chromosome 18. Translocation of this protooncogene to chromosome 14 in proximity to the immunoglobulin heavy chain gene leads to overexpression of bcl-2, which decreases apoptosis and confers a survival advantage to the cell. Studies show that p53 is also a regulator of apoptosis. Loss of p53 disrupts normal apoptotic pathways, imparting a survival advantage to the cell. Apoptosis may also play an important role as a mechanism of inherent resistance to chemotherapy.11
When the normal regulatory mechanisms for cellular growth fail, backup defense systems may be activated. The secondary defenses include apoptosis (programmed cell death or suicide) and cellular senescence (aging). Apoptosis is a normal mechanism of cell death required for tissue homeostasis.3,7,11 This process is regulated by oncogenes and tumor suppressor genes and is also a mechanism of cellular death after exposure to cytotoxins. Overexpression of oncogenes responsible for apoptosis may produce an “immortal” cell, which has increased potential for malignancy. The bcl-2 oncogene is an example. The most common chromosomal abnormality found in lymphoid malignancies is the t(14;18) translocation. The bcl-2 protooncogene is normally located on chromosome 18. Translocation of this protooncogene to chromosome 14 in proximity to the immunoglobulin heavy chain gene leads to overexpression of bcl-2, which decreases apoptosis and confers a survival advantage to the cell. Studies show that p53 is also a regulator of apoptosis. Loss of p53 disrupts normal apoptotic pathways, imparting a survival advantage to the cell. Apoptosis may also play an important role as a mechanism of inherent resistance to chemotherapy.11
Cellular senescence is another important defense mechanism.6,7 Laboratory studies demonstrate that after a cell population has undergone a preset number of doublings, growth stops, and cells die. This is known as senescence, a process that is regulated by telomeres. Telomeres are the DNA segments or caps at the ends of chromosomes. They are responsible for protecting the end of the DNA from damage. With each replication, the length of the telomeres is shortened. After the telomeres are shortened to a critical length, senescence is triggered. In this way, telomeres tally and limit the number of cell doublings. In cancer cells, the function of telomeres is overcome by overexpression of an enzyme known as telomerase. Telomerase replaces the portion of the telomeres that is lost with each cell division, thereby avoiding senescence and permitting an infinite number of cell doublings. Telomerase is a target for anticancer agent development.
![]() As information regarding the role of oncogenes and tumor suppressor genes accumulated, it became evident that a single mutation is probably insufficient to initiate cancer.4–7 Scientists postulate that combinations of mutations are required for carcinogenesis and that each mutation is inherited by the next generation of cells. Thus, several detectable genetic mutations may be present in an established tumor. Whereas early mutations are found in both premalignant lesions and established tumors, later mutations are found only in the established tumor. This theory of sequential genetic mutations resulting in cancer has been demonstrated in colon cancer. In colon cancer, the initial genetic mutation is believed to be loss of the adenomatous polyposis coli gene, which results in formation of a small benign polyp. Oncogenic mutation of the ras gene is often the next step, leading to enlargement of the polyp. Loss of function of DNA mismatch repair enzymes may occur at many points in the progression of malignant transformation. Loss of the p53 gene and another gene, believed to be the deleted in colorectal cancer (DCC) gene, completes the transformation into a malignant lesion. Loss of p53 is thought to be a late event in the development and progression of the malignancy.
As information regarding the role of oncogenes and tumor suppressor genes accumulated, it became evident that a single mutation is probably insufficient to initiate cancer.4–7 Scientists postulate that combinations of mutations are required for carcinogenesis and that each mutation is inherited by the next generation of cells. Thus, several detectable genetic mutations may be present in an established tumor. Whereas early mutations are found in both premalignant lesions and established tumors, later mutations are found only in the established tumor. This theory of sequential genetic mutations resulting in cancer has been demonstrated in colon cancer. In colon cancer, the initial genetic mutation is believed to be loss of the adenomatous polyposis coli gene, which results in formation of a small benign polyp. Oncogenic mutation of the ras gene is often the next step, leading to enlargement of the polyp. Loss of function of DNA mismatch repair enzymes may occur at many points in the progression of malignant transformation. Loss of the p53 gene and another gene, believed to be the deleted in colorectal cancer (DCC) gene, completes the transformation into a malignant lesion. Loss of p53 is thought to be a late event in the development and progression of the malignancy.
Identification of genes and other proteins involved in carcinogenesis has several important clinical implications. They may be used in cancer screening to identify individuals at increased risk for cancer and are being used to design new anticancer agents and gene therapies, several of which have recently been approved for use. Specific genetic abnormalities are so commonly associated with some cancers that the presence of that abnormality may aid in the diagnosis of that cancer. If the presence of these genes (i.e., gene expression profile) can reliably predict the clinical course of a cancer or response to certain cancer therapies, then genetic analysis may also become an important prognostic and treatment decision tool. An example of this is overexpression of HER2 predicting response to trastuzumab.9
Oncogenes and Tumor Suppressor Genes
Recent advances in molecular biology have identified many oncogenes and tumor suppressor genes that contribute to the functional capabilities that are acquired by cancer cells. These activated oncogenes and mutated tumor suppressor genes can be segregated into families and identified by their interactions with specific intracellular signaling pathways. Monoclonal antibodies (MoABs) have been developed to target the extracellular receptors or their natural ligands and prevent ligand binding to the receptor. In addition, small molecular inhibitors that target intracellular tyrosine kinases receptors or signal transduction pathways are available. The net effect of both strategies is to prevent downstream activation of the signal transduction resulting in a decrease in cell proliferation (Fig. 104-3). Some common receptors and pathways targeted by available targeted drugs and MoABs include HER, vascular endothelial growth factor (VEGF), mitogen-activated protein kinase pathway (MAPK) and phosphatidylinositide 3-kinase (PI3K) pathway.
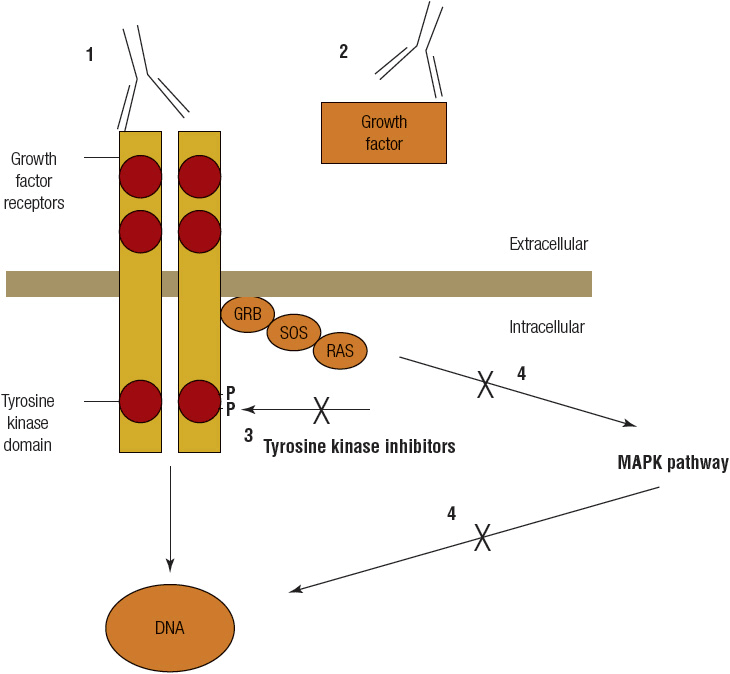
FIGURE 104-3 Common elements of intracellular signaling pathways and targeted strategies that inhibit these pathways, such as (1) monoclonal antibodies (MoABs) against the growth factor receptor, (2) MoABs against the growth factor itself, (3) molecules that target intracellular tyrosine kinases and prevent phosphorylation of tyrosine residues and subsequent activation of downstream signals, and (4) targeting downstream signals. All targeted therapies have the same goal of decreasing cell proliferation and increasing cell death of cancer cells. (MAPK, mitogen-activated protein kinase.)
Human Epidermal Growth Factor Receptors
![]() Targeting the HER pathway is currently used to treat a variety of solid tumors. The HER family of receptors contains four known members, which upon binding to growth factor ligands result in intracellular phosphorylation of transcription factors and cell proliferation (Fig. 104-4).9,12,13 EGFR and HER2 are known to be overexpressed in several cancers, including breast, lung, gastric, and colon cancers. Activation of these receptors leads to uncontrolled cellular growth and proliferation, tumor metastasis, and the prevention of apoptosis in cancer cells.12 The roles of HER3 and HER4 in cancer growth and proliferation are still under investigation. All members of this family contain a transmembrane glycoprotein extracellular ligand binding site, a transmembrane domain, and a cytosolic tyrosine kinase tail. Members of the HER family are inactive by themselves and must form a dimer (a molecule composed of two subunits) either with a member of the same family (homodimer) or with a member of a different HER family (heterodimer).22 Dimerization of the receptor leads to tyrosine kinase phosphorylation and subsequent activation of downstream pathways required to activate signal transduction and cell growth.
Targeting the HER pathway is currently used to treat a variety of solid tumors. The HER family of receptors contains four known members, which upon binding to growth factor ligands result in intracellular phosphorylation of transcription factors and cell proliferation (Fig. 104-4).9,12,13 EGFR and HER2 are known to be overexpressed in several cancers, including breast, lung, gastric, and colon cancers. Activation of these receptors leads to uncontrolled cellular growth and proliferation, tumor metastasis, and the prevention of apoptosis in cancer cells.12 The roles of HER3 and HER4 in cancer growth and proliferation are still under investigation. All members of this family contain a transmembrane glycoprotein extracellular ligand binding site, a transmembrane domain, and a cytosolic tyrosine kinase tail. Members of the HER family are inactive by themselves and must form a dimer (a molecule composed of two subunits) either with a member of the same family (homodimer) or with a member of a different HER family (heterodimer).22 Dimerization of the receptor leads to tyrosine kinase phosphorylation and subsequent activation of downstream pathways required to activate signal transduction and cell growth.
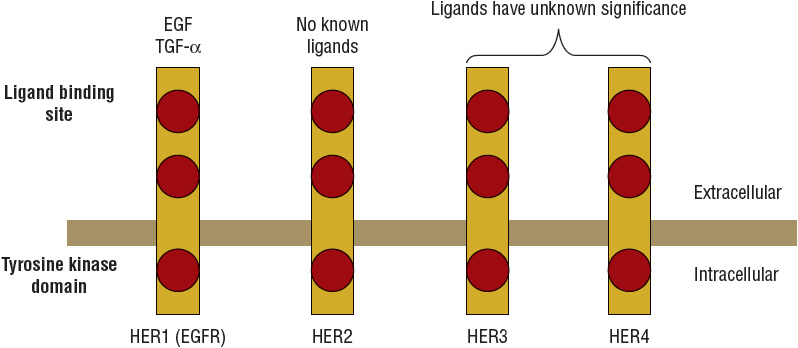
FIGURE 104-4 The human epidermal growth factor receptor (HER) family of growth factor receptors. All members of the HER family contain a transmembrane glycoprotein, an extracellular ligand binding site, and a hydrophobic intracellular portion with a tyrosine kinase domain. HER1 (or more commonly called EGFR [epidermal growth factor receptor]) has several known ligands, and HER2 has no known ligands, but the significance of ligands for HER3 and HER4 is unknown at this time. After the molecule binds to another member of the HER family, the tyrosine kinase domain is phosphorylated, and genes regulating proliferation, antiapoptosis, and cell transformation are turned on. (TGF, transforming growth factor.)
Vascular Endothelial Growth Factor
![]() Vascular endothelial growth factor (VEGF) is a protein that stimulates angiogenesis, which is the development of new blood vessels. Angiogenesis is a process important for normal physiologic processes, but becomes unregulated in several malignancies and can lead to tumor growth, invasion, and metastasis. This process is regulated by pro- and antiangiogenic growth factors, which are released in response to hypoxia and other stresses to the cell.14 Proangiogenic growth factors include VEGF, fibroblast growth factors, platelet-derived growth factor (PDGF), tumor necrosis factor-α (TNF-α), and keratinocyte growth factor. Antiangiogenic growth factors include interleukin-12 (IL-12), interferons (IFNs), platelet factor 4, and tissue inhibitors of metalloproteinase.15
Vascular endothelial growth factor (VEGF) is a protein that stimulates angiogenesis, which is the development of new blood vessels. Angiogenesis is a process important for normal physiologic processes, but becomes unregulated in several malignancies and can lead to tumor growth, invasion, and metastasis. This process is regulated by pro- and antiangiogenic growth factors, which are released in response to hypoxia and other stresses to the cell.14 Proangiogenic growth factors include VEGF, fibroblast growth factors, platelet-derived growth factor (PDGF), tumor necrosis factor-α (TNF-α), and keratinocyte growth factor. Antiangiogenic growth factors include interleukin-12 (IL-12), interferons (IFNs), platelet factor 4, and tissue inhibitors of metalloproteinase.15
The best studied proangiogenic factor is VEGF, whose elevated levels have been associated with a poor prognosis and an increased risk of metastases in a variety of malignancies, including acute myelogenous leukemia (AML), breast cancer, hepatocellular carcinoma, non–small cell lung cancer (NSCLC), ovarian cancer, and colon cancer.15 Similar to other growth factors, VEGF binds to specific receptors located on the extracellular domain of growth factor receptors. Three known receptors of VEGF have been identified: VEGFR-1, -2, and -3.15 The VEGFR-1 and VEGFR-2 receptors are expressed primarily in endothelial cells and in some cancer cells and mediate the biologic effects of VEGF. Each of the receptors induces a different signal transduction pathway. These pathways eventually result in the generation of proteases that are necessary for the breakdown of the extracellular matrix, the first step of angiogenesis. Interference with their ability to develop new blood vessels by means of antiangiogenic agents can limit or prevent tumor growth.15
Intracellular Signaling Pathways
Well-described intracellular signaling pathways include PI3K, JAK-STAT (Janus kinase–signal transducers and activators of transcription), and MAPK pathways; when activated, they promote cell proliferation and survival. These pathways consist of a chain of proteins that ultimately communicate a signal to the DNA found in the nucleus from a cell surface receptor (e.g., EGFR). A protein within a signaling pathway communicates by adding a phosphate group to its neighboring protein; the phosphate groups act as “on” or “off” switch for the pathway. In cancer, a mutated protein permits the pathway to remain in the “on” or “off” position. The downstream effectors of these pathways also initiate cell cycle progression by promoting the expression of cyclins and repressing the expression of CDK inhibitors.
The MAPK signaling pathways regulates many fundamental cellular processes, including cell differentiation, proliferation, and senescence. These pathways relay the intracellular signals through a series of Ras, Raf, MEK (MAPK/ERK) and ERK (extracellular signaling receptor kinase) proteins that subsequently phosphorylate and regulate nuclear and cytoplasmic structures. Some of these proteins are commonly mutated in pancreatic, melanoma, colorectal, hepatocellular, and other solid tumors.16
The PI3K signaling pathway also regulates cell proliferation, growth, survival, and mobility. PI3K becomes activated in response to growth hormones, and it ultimately activates AKT, a serine–threonine kinase that serves as a master switch for the cell cycle progression. Fully activated AKT translocates to the nucleus, where it can inhibit proapoptotic signals and activate antiapoptotic substrates. It can also phosphorylate mammalian target of rapamycin (mTOR). After being activated, mTOR stimulates protein synthesis by phosphorylating translation regulators. mTOR also contributes to protein degradation and angiogenesis.17 Phosphatase and tensin homolog (PTEN) is a tumor suppressor gene that blocks intracellular signaling through this pathway and is frequently inactivated in several solid tumors.18,19
The JAK-STAT signaling pathway helps regulate the immune system. This pathway contains three main components: extracellular receptors, JAKs, and STAT. The pathway is initiated when cytokines or growth factors bind to the receptor, activate JAK, and subsequently recruit STAT. The STAT proteins then translocate to the nucleus and modify gene expression. Altered JAK signaling has been associated with JAK mutations in patients with myelofibrosis.20
Epigenetics
Epigenetics refers to changes in gene expression that occur without altering the DNA sequence.21 The two most common mechanisms of epigenetic regulation include methylation and histone modification. DNA methylation commonly occurs at CpG dinucleotides (or islands) and is catalyzed by DNA methyltransferases (DNMT). Histones are basic proteins associated with DNA in the nucleosome. These proteins may be modified by acetylation, methylation, or phosphorylation on their N-terminal tail. These modifications play a role in transcriptional regulation. For example, whereas histone deacetylases (HDACs) repress transcription, histone acetylases activate transcription. Epigenetic changes may be involved in the development of cancer by either priming the cell and making it susceptible to genetic changes associated with the development of cancer or initiating malignant transformation. As an example, hypermethylation at CpG dinucleotides found near tumor suppressor genes can switch these genes off and promote the development of cancer. Anticancer agents, identified as inhibitors of DNMT or HDAC, target these modifications. Figure 104-5 shows the effects of these inhibitors on methylation, chromatin formation, and transcription.
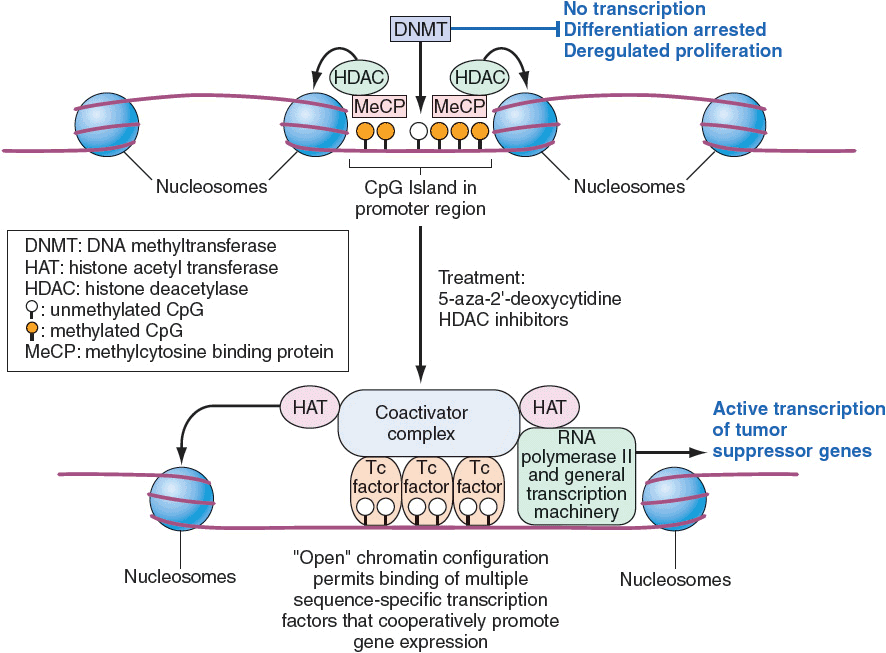
FIGURE 104-5 Epigenetic regulation of gene expression in cancer cells. CpG islands within the promoter and enhancer regions of the gene are methylated, resulting in the complexes with histone deacetylase (HDAC) activity. Chromatin is in a condensed conformation that inhibits transcription (upper figure). Inhibitors of DNMT in combination with inhibitors of HDAC confer a chromatin structure that allows transcription (lower figure). (From Longo DL. Cancer Cell Biology and Angiogenesis. In: Longo DL, Fauci AS, Kasper DL, et al. eds. Harrison’s Principles of Internal Medicine, 18th ed. New York, NY: McGraw-Hill, 2012.)
PATHOLOGY OF CANCER
Tumor Origin
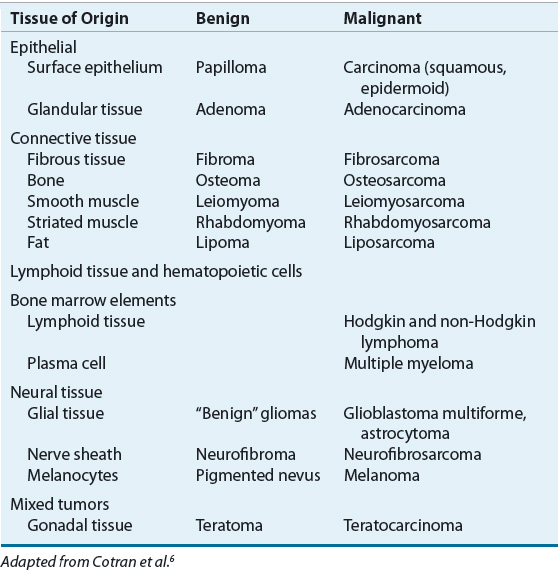
Tumors may arise from any of four basic tissue types: epithelial tissue, connective tissue (i.e., muscle, bone, and cartilage), lymphoid tissue, and nerve tissue. Most cancer cells retain enough traits to identify their basic tissue type; therefore, tumors are typically named based on the tissue of origin. For example, benign tumors are named for their cell or tissue of origin followed the suffix –oma. Table 104-3 lists common tumor nomenclature by tissue type.6
TABLE 104-3 Tumor Classification by Tissue Type
Some cancers are preceded by cellular changes that are abnormal but not yet malignant. Correcting these early changes could potentially prevent the occurrence of a cancer. These precancerous lesions may be described as consisting of either hyperplastic or dysplastic cells. Hyperplasia is an increase in the number of cells in a particular tissue or organ, which results in an increased size of the organ. It should not be confused with hypertrophy, which is an increase in the size of the individual cells. Hyperplasia occurs in response to a stimulus and reverses when the stimulus is removed. Dysplasia is defined as an abnormal change in the size, shape, or organization of cells or tissues. Hyperplasia and dysplasia may precede the appearance of a cancer by several months or years.
Cancer cells are divided into those of epithelial origin or the other tissue types. Carcinomas are malignant growths arising from epithelial cells and sarcomas are malignant growths of muscle or connective tissue. Carcinoma in situ is a preinvasive stage of malignancy in which the cancer is limited to the epithelial cells or origin. Malignancies of hematologic origin, such as leukemias and lymphomas, are classified separately. Leukemias and lymphomas are discussed in later chapters.
Tumor Characteristics
Tumors may be either benign or malignant. Benign tumors are noncancerous growths that are often encapsulated, localized, and indolent. The cells of benign tumors resemble the cells from which they developed. These masses seldom metastasize, and after being removed, they rarely recur. In contrast, malignant tumors invade and destroy the surrounding tissue. The cells of malignant tumors are genetically unstable, and loss of normal cell architecture results in cells that are atypical of their tissue or cell of origin. These cells lose the ability to perform their usual functions. This loss of structure and function is called anaplasia. Malignant tumors tend to metastasize, and consequently, recurrences are common after removal or destruction of the primary tumor.
Invasion and Metastasis
![]() Metastasis is the spread of cancer cells from the primary tumor site to distant sites.5,22 Despite advances in diagnostic techniques and screening for cancer, many patients have metastatic disease at diagnosis. When distant metastases are clinically evident, cancers are seldom curable. Newly diagnosed cancer patients may also have microscopic cancer metastases (i.e., micrometastases). Although clinically undetectable, these microscopic metastases must be present because many patients subsequently relapse at distant sites despite removal or destruction of the primary tumor. However, some patients with micro-metastatic disease may be cured with systemic therapy.
Metastasis is the spread of cancer cells from the primary tumor site to distant sites.5,22 Despite advances in diagnostic techniques and screening for cancer, many patients have metastatic disease at diagnosis. When distant metastases are clinically evident, cancers are seldom curable. Newly diagnosed cancer patients may also have microscopic cancer metastases (i.e., micrometastases). Although clinically undetectable, these microscopic metastases must be present because many patients subsequently relapse at distant sites despite removal or destruction of the primary tumor. However, some patients with micro-metastatic disease may be cured with systemic therapy.
The two primary pathways of metastasis are hematogenous and lymphatic. Other less common modes of disease spread include dissemination via cerebrospinal fluid and transabdominal spread within the peritoneal cavity. Tumors constantly shed neoplastic cells into the systemic circulation or surrounding lymphatics. The onset and time course for the development of metastasis depends largely on the tumor biology. Breast cancer, for example, tends to metastasize very early. Not all of the shed cancer cells result in a metastatic lesion; the cells must first find an environment suitable for growth.22 This process is illustrated in the diverse patterns of metastasis observed for different cancers. As an example, prostate cancer commonly metastasizes to bone but rarely to the brain.
The process of invasion and metastasis involves several essential steps. After transformation, the cancer cells and surrounding host tissue secrete substances that stimulate angiogenesis.23 Cancer cells must then detach from the primary mass and invade surrounding blood and lymph vessels. The cancer cells or cell aggregates detach and embolize through these vessels, but most do not survive circulation. The disseminated cells must then attach to the vascular endothelium. The cells may proliferate within the lumen of the vessel but most commonly extravasate into the surrounding tissue. The local microenvironment may provide growth factors that can serve as “fertilizer” to potentiate the proliferation of the metastasis. At every step, the potential metastatic cell must fight the host immune system. Finally, the metastasis must again initiate angiogenesis to ensure continued growth and proliferation. Because angiogenesis has been recognized as a critical element in primary tumor growth as well as metastasis, it has become a target for development of new anticancer agents, which are described later in the chapter.
DIAGNOSIS AND STAGING
Screening
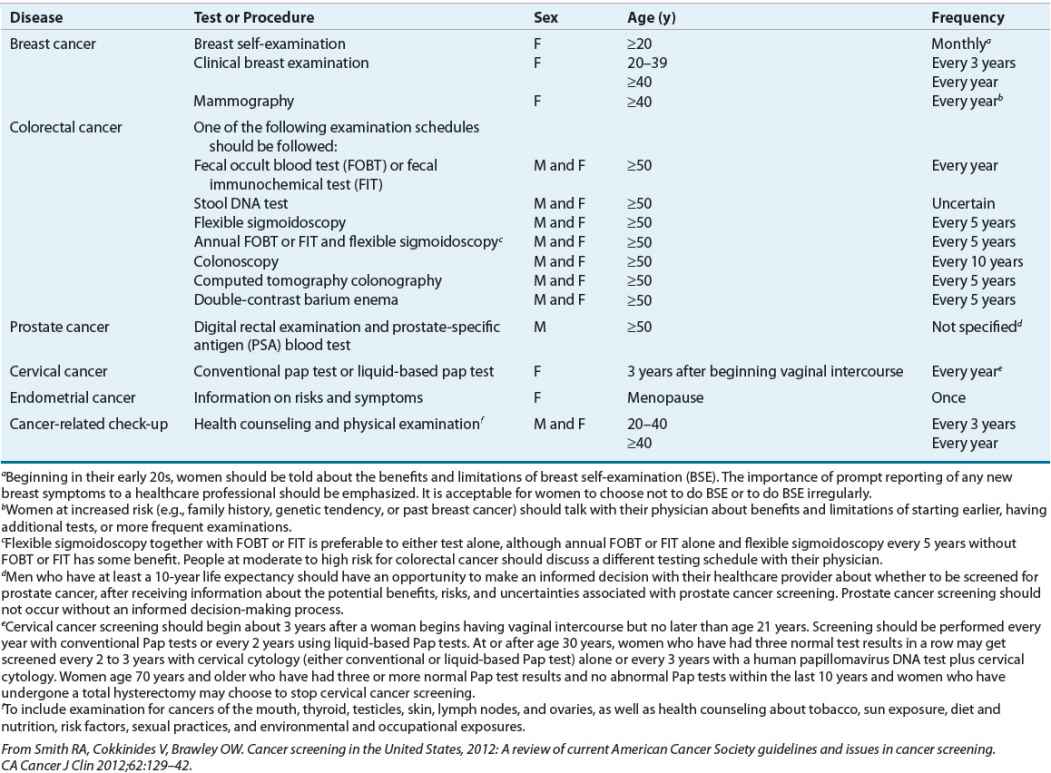
Because cancers are most curable before they have metastasized, early detection and treatment have obvious potential benefits. Cancer screening programs are designed to detect signs of cancer in people who have not yet developed symptoms from cancer. Lack of effective screening methods and inaccessible anatomic sites limit the availability of screening methods for some cancers. Other limitations of screening methods include false-negative test results (related to the sensitivity of the test), false-positive test result (related to the specificity), and overdiagnosis (true positives that will not become clinically significant). For example, most abnormal test results identified by a screening mammography are false-positive results, although the specificity of this screening method exceeds 90%. Public education on the early warning signs of common cancers is extremely important for facilitating early detection. Effective screening procedures exist for some cancers. The Papanicolaou (Pap) smear test, for example, is an effective tool to detect cervical cancer in its early stages. The American Cancer Society publishes yearly guidelines for routine screening examinations (Table 104-4).24
TABLE 104-4 Screening Guidelines for Early Detection of Cancer in Average-Risk, Asymptomatic People
Diagnosis
The presenting signs and symptoms of cancer vary widely and depend on the cancer. The presentation in adults may include any of cancer’s seven warning signs (Table 104-5), pain, or loss of appetite.25 The warning signs of cancer in pediatrics are different and reflect the tumors more common in this population (Table 104-6).25 Even with increased public awareness, the fear of a cancer diagnosis can deter people from seeking medical attention. The definitive diagnosis of cancer relies on the procurement of a sample of the tissue or cells suspected of malignancy and pathologic assessment of this sample. This sample can be obtained by numerous methods, including biopsy, exfoliative cytology, or fine-needle aspiration. A tissue diagnosis is essential, because many benign conditions can masquerade as cancer. Definitive treatment should not begin without a pathologic diagnosis.
TABLE 104-5 Cancer’s Seven Warning Signs

TABLE 104-6 Cancer’s Warning Signs in Children

Staging and Workup
![]() In addition to tissue diagnosis, tumors should be staged to determine the extent of disease before any definitive treatment is initiated. The process is dictated by knowledge of the biology of the tumor and by the signs and symptoms elicited in the history and physical examination. Staging provides information on prognosis and guides treatment selection. A staging workup may involve radiographs, computed tomography scans, magnetic resonance imaging, positron emission tomography scans, ultrasonograms, bone marrow biopsies, bone scans, lumbar puncture, and a variety of laboratory tests (including appropriate tumor markers). After treatment is implemented, the staging workup is usually repeated to evaluate the effectiveness of the treatment. Some cancers produce antigens or other substances; these tumor markers are often nonspecific and may be elevated in many different cancers or in patients with nonmalignant diseases. As a result, tumor markers are generally more useful for monitoring response and detecting recurrence than as diagnostic tools. Examples are human chorionic gonadotropin and alfa-fetoprotein in patients with testicular cancer or prostate-specific antigen in prostate cancer.6
In addition to tissue diagnosis, tumors should be staged to determine the extent of disease before any definitive treatment is initiated. The process is dictated by knowledge of the biology of the tumor and by the signs and symptoms elicited in the history and physical examination. Staging provides information on prognosis and guides treatment selection. A staging workup may involve radiographs, computed tomography scans, magnetic resonance imaging, positron emission tomography scans, ultrasonograms, bone marrow biopsies, bone scans, lumbar puncture, and a variety of laboratory tests (including appropriate tumor markers). After treatment is implemented, the staging workup is usually repeated to evaluate the effectiveness of the treatment. Some cancers produce antigens or other substances; these tumor markers are often nonspecific and may be elevated in many different cancers or in patients with nonmalignant diseases. As a result, tumor markers are generally more useful for monitoring response and detecting recurrence than as diagnostic tools. Examples are human chorionic gonadotropin and alfa-fetoprotein in patients with testicular cancer or prostate-specific antigen in prostate cancer.6
The most commonly applied staging system for solid tumors is the TNM classification, where T = tumor, N = node, and M = metastases. A numerical value is assigned to each letter to indicate the size or extent of disease. The designated rating for a tumor describes the size of the primary mass and ranges from T1 to T4. Carcinoma in situ is designated as Tis. Nodes are described in terms of the extent of the spread of regional lymph nodal involvement (N0 to N3). Metastases are generally scored depending on their presence or absence (M0 or M1). To simplify the staging process, most cancers are classified according to the extent of disease by a numerical system involving stages I through IV. Stage I usually indicates localized tumor, stages II and III represent local and regional spread of disease, and stage IV denotes the presence of distant metastases. The assigned TNM rating translates into a particular stage classification. For example, T3 N1 M0 describes a moderate- to large-sized primary mass, with regional lymph node involvement and no distant metastases and for most cancers is stage III. The criteria for classifying disease extent are quite specific for each different cancer.26 For some tumors, such as prostate cancer, alternative alphabetical systems (stage A, B, C, or D) are used in clinical practice. Leukemias and lymphomas follow alternate staging systems that are discussed in subsequent chapters.
TREATMENT
Modalities of Cancer
Five primary modalities are used to treat cancer: surgery, radiation, traditional chemotherapy, targeted drug therapy, and biologic therapy. The oldest treatment modality is surgery, which plays a major role in the diagnosis and treatment of cancer. Surgery remains the treatment of choice for most solid tumors diagnosed in the early stages. Radiation therapy was first used for cancer treatment in the late 1800s and remains a mainstay in the management of cancer. Although very effective for treating many cancers, surgery and radiation are local treatments. These modalities are likely to produce a cure in patients with truly localized disease. Because most patients with cancer have micrometastatic or metastatic disease at diagnosis, localized anticancer treatment often fail to completely eliminate the cancer. In addition, systemic diseases such as leukemia cannot be treated with a localized modality. Chemotherapy, targeted drug therapies, and biologic therapies all access the systemic circulation and can theoretically treat the primary tumor or metastatic disease. Biologic therapies are made from a living organism or its products and include antibodies, vaccines, growth factors, and cytokines.
![]() Many solid tumors or lymphomas appear to be eliminated by surgery or radiation. However, the high incidence of disease recurrence implies that the primary tumor metastasized before it was removed. Adjuvant therapy is the use of systemic therapy to eradicate micrometastatic disease after localized modalities, such as surgery or radiation. The goal of adjuvant therapy is to reduce recurrence rates and prolong long-term survival. Thus, adjuvant therapy is given to patients with potentially curable malignancies who have no clinically detectable disease after surgery or radiation. Because adjuvant therapy is given at a time when the cancer is undetectable (i.e., no measurable disease), its effectiveness is evaluated by recurrence rates and survival. The value of adjuvant therapy is best established in colorectal and breast cancers. Systemic therapy may also be given in the neoadjuvant or preoperative setting. The goals in these instances are to make other treatment modalities more effective by reducing tumor burden and destroying micrometastases. For example, in breast cancer, it is often used to reduce the size of the primary tumor and allow for a less invasive surgical procedure.
Many solid tumors or lymphomas appear to be eliminated by surgery or radiation. However, the high incidence of disease recurrence implies that the primary tumor metastasized before it was removed. Adjuvant therapy is the use of systemic therapy to eradicate micrometastatic disease after localized modalities, such as surgery or radiation. The goal of adjuvant therapy is to reduce recurrence rates and prolong long-term survival. Thus, adjuvant therapy is given to patients with potentially curable malignancies who have no clinically detectable disease after surgery or radiation. Because adjuvant therapy is given at a time when the cancer is undetectable (i.e., no measurable disease), its effectiveness is evaluated by recurrence rates and survival. The value of adjuvant therapy is best established in colorectal and breast cancers. Systemic therapy may also be given in the neoadjuvant or preoperative setting. The goals in these instances are to make other treatment modalities more effective by reducing tumor burden and destroying micrometastases. For example, in breast cancer, it is often used to reduce the size of the primary tumor and allow for a less invasive surgical procedure.
These modalities may be used alone, but are typically used in combination. Early-stage breast cancer is a good example of the use of a combined-modality approach. The primary tumor is removed surgically, and radiation therapy is delivered to the remaining breast (after lumpectomy) or to the axilla (if there is marked lymph node involvement). Adjuvant therapy, including chemotherapy and biologic therapy, is then administered to eradicate any micrometastatic disease. Neoadjuvant therapy may sometimes be administered before definitive surgery to increase the likelihood of a tumor resection compared with a mastectomy.
The management of hematologic malignancies also involves the use of combined modalities, but the terminology is different. Chemotherapy that is administered to eradicate the cancer cells is called induction therapy. When a complete remission (the disappearance of all signs of the cancer) is documented, postremission or consolidation therapy is administered. These therapies are designed to eradicate any remaining disease, similar to adjuvant therapy for solid tumors, and can include systemic therapy, a hematopoietic stem cell transplant, or radiation therapy. Maintenance therapy is sometimes administered after consolidation therapy. This therapy is given to prevent the cancer from recurring and may include combination chemotherapy.
When an anticancer agent is administered to patients with local or regional disease, the treatment is often administered to cure the patient and may be labeled curative therapy. However, when the cancer has metastasized to distant sites, cure is usually not possible. Anticancer agents can be administered to patients with metastatic disease to slow the progression of cancer and prolong survival by months to years. Anticancer agents administered to patients with terminal cancer with the goal of reducing symptoms is called palliative therapy.
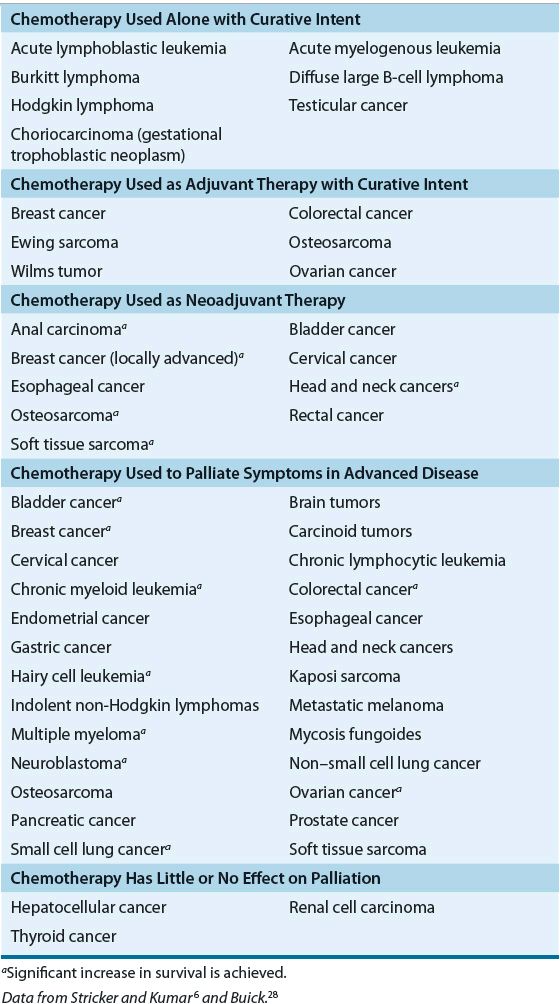
The era of modern cancer chemotherapy was born in 1941 when Goodman and Gilman first administered nitrogen mustard to patients with lymphoma.27 Since then, numerous anticancer agents have been developed, and a variety of treatment regimens have been investigated in every cancer. Table 104-7 lists tumors and their responsiveness to chemotherapy.6,28 Chemotherapy may be indicated as a curative or palliative. Treatment with chemotherapy is the primary curative modality for a few diseases, including leukemias, lymphomas, choriocarcinomas, and testicular cancer. Most solid tumors are not curable with chemotherapy alone, either because of the biology of the tumor or because of advanced disease at presentation. Chemotherapy in this setting is often initiated for palliative purposes. It is often possible to decrease tumor size or to retard growth enough to reduce untoward symptoms caused by the tumor.
TABLE 104-7 The Role of Chemotherapy in the Treatment of Cancer
MOLECULAR AND CELLULAR BASIS
Principles of Tumor Growth
The study of tumor growth forms the foundation for many of the basic principles of modern cancer chemotherapy. The growth of most tumors is illustrated by the gompertzian tumor growth curve (Fig. 104-5).6,28,29 Gompertz was an insurance actuary who described the relationship between age and expected death. This mathematical model also approximates tumor cell proliferation. In the early stages, tumor growth is exponential, which means that the tumor takes a constant amount of time to double its size. During this early phase, most cancer cells are actively dividing. This population of cells is called the growth fraction. The doubling time, or time required for the tumor to double in size, is very short. Because most anticancer agents have greater effect on rapidly dividing cells, tumors are most sensitive to their effects when the tumor is small and the growth fraction is high. However, as the tumor grows, the doubling time is slowed.28,29 The growth fraction is decreased, probably owing to the tumor’s outgrowing its blood and nutrient supply or the inability of blood and nutrients to diffuse throughout the tumor mass. Wide variability exists in measured doubling times for different cancers. The doubling time of most solid tumors is about 2 to 3 months. However, some tumors have doubling times of only days (e.g., aggressive non-Hodgkin lymphomas [NHLs]), and others have even longer doubling times (e.g., some salivary gland tumors).6
Figure 104-6 also illustrates the impact of tumor burden. It takes about 109 cancer cells (1-g mass, 1 cm in diameter) for a tumor to be clinically detectable by palpation or radiography. Such a tumor has undergone about 30 doublings in cell number. It only takes 10 additional doublings for this 1-g mass to reach 1 kg in size. A tumor possessing 1012 cells (1-kg mass) is considered lethal. Thus, a tumor is clinically undetectable for most of its life span. Tumor burden also impacts response to treatment. The cell kill hypothesis states that a certain percentage of cells (not a certain number of cells) will be killed with each treatment course. For example, if a tumor consists of 1,000 cells and the treatment kills 90% of the cells, then 10% or 100 cells remain. The second treatment course kills another 90% of cells, and again only 10% or 10 cells remain. According to this hypothesis, the tumor burden will never reach zero. Tumors consisting of less than 104 cells are believed to be small enough for elimination by host factors, including immunologic mechanisms, and these factors must be in place for a cure to be possible. The limitations of this theory are that it assumes all cancers are equally responsive and that resistance to anticancer agents and metastases do not occur.1,6,28,29
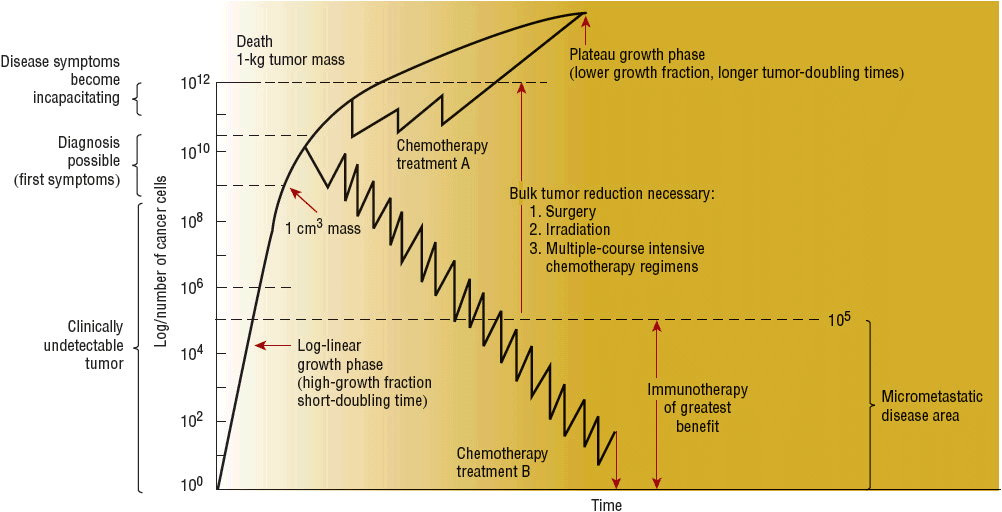
FIGURE 104-6 Gompertzian kinetics tumor-growth curve: relationship to symptoms, diagnosis, and various treatment regimens. (Reproduced with permission from Buick RN. Cellular basis of chemotherapy. In: Dorr RT, Von Hoff DD, eds. Cancer Chemotherapy Handbook, 2nd ed. New York: Appleton & Lange/McGraw-Hill, 1994:3–14.)
Tumor Proliferation
Both cancer cells and normal cells reproduce in a series of steps known as the cell cycle as described earlier in the chapter. Figure 104-7 depicts the cell cycle and the phases of activity for some traditional chemotherapies.28,29
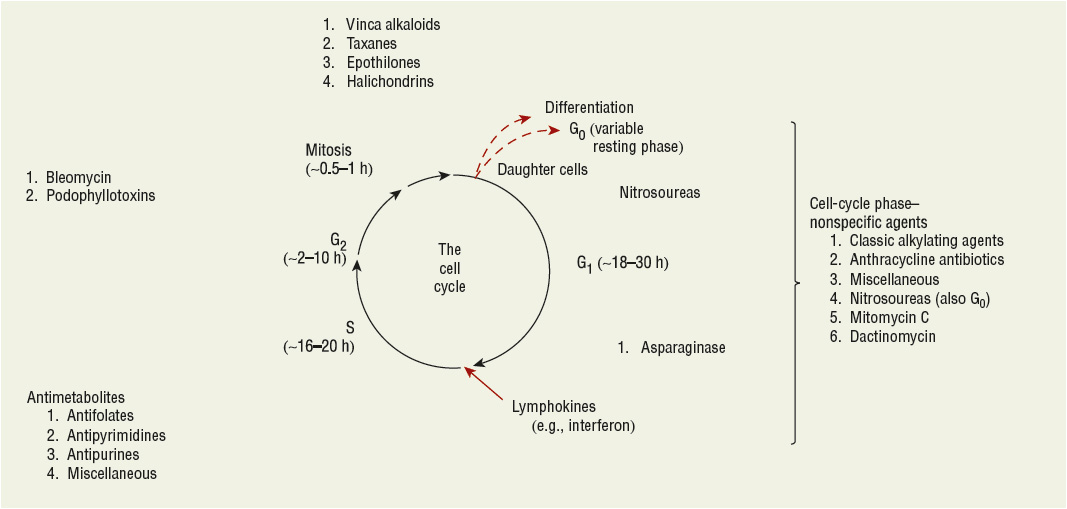
FIGURE 104-7 Cell-cycle activity for anticancer drugs. Cell-cycle phase-specific chemotherapies appear to be most active during a particular phase but may also be active in another phase. Cell-cycle phase-nonspecific chemotherapies may have greater activity in one phase than another but not to the degree of cell-cycle phase-specific chemotherapies. In many cases, it is likely that chemotherapy cytotoxicity involves multiple intracellular sites of action and may not be linked to specific cell-cycle events.
![]() All cancer cells do not proliferate faster than normal cells; some cancer cells reproduce more rapidly, but others are more indolent. Many chemotherapies target rapidly proliferating cells (both normal and cancerous cells), and these therapies might act at selective or multiple sites of the cell cycle. Chemotherapy that demonstrates major activity in a particular phase of the cell cycle are known as cell-cycle phase-specific chemotherapies. For example, antimetabolites exert their major effect during the S phase. Cell-cycle phase-specific therapies may also be active to a lesser extent in other phases of the cycle. Cell-cycle phase-nonspecific chemotherapy are those with significant activity in multiple phases. Alkylating agents, such as nitrogen mustards, are examples of a cell-cycle nonspecific agent. In many cases, the cytotoxic effects of an agent may result from interactions with other intracellular activities and are not related to specific cell-cycle events. Endocrine therapies and targeted drugs are examples of these anticancer agents.
All cancer cells do not proliferate faster than normal cells; some cancer cells reproduce more rapidly, but others are more indolent. Many chemotherapies target rapidly proliferating cells (both normal and cancerous cells), and these therapies might act at selective or multiple sites of the cell cycle. Chemotherapy that demonstrates major activity in a particular phase of the cell cycle are known as cell-cycle phase-specific chemotherapies. For example, antimetabolites exert their major effect during the S phase. Cell-cycle phase-specific therapies may also be active to a lesser extent in other phases of the cycle. Cell-cycle phase-nonspecific chemotherapy are those with significant activity in multiple phases. Alkylating agents, such as nitrogen mustards, are examples of a cell-cycle nonspecific agent. In many cases, the cytotoxic effects of an agent may result from interactions with other intracellular activities and are not related to specific cell-cycle events. Endocrine therapies and targeted drugs are examples of these anticancer agents.
Knowledge of cell-cycle specificity has been use to optimize the scheduling of chemotherapy. By definition, cell-cycle phase-specific chemotherapies exert their major activity when cells are in a particular phase of the cell cycle. At any given time, the heterogeneous cell populations within a tumor are at various phases in the cell cycle. By giving cell-cycle phase-specific chemotherapies as a continuous infusion or in multiple repeated fractions, healthcare providers can theoretically target more cells as they progress into the sensitive phase. Thus, cell-cycle phase-specific chemotherapies are also termed schedule dependent. In contrast, cell-cycle phase-nonspecific chemotherapies are active in many phases and consequently are not schedule dependent. The activity of these chemotherapies depends on the dose and these chemotherapies are termed dose dependent.
Biologic therapies and targeted drugs interfere with cancer cell proliferation in a different manner compared with traditional chemotherapy. These therapies stop cancer progression by blocking aberrant intracellular signaling pathways that govern cell responses, movement, and division. Some of these agents can cause cancer cell death by inducing apoptosis or stimulating the immune system to destroy the cancer cells. Some targeted drug therapies and biologic therapies are used in combination with traditional chemotherapy.
Molecular Biology
Because many anticancer agents interfere with the cellular synthesis of DNA, RNA, and proteins, it is important to review the basic principles of molecular biology.3 Each normal human cell contains 46 chromosomes, which are composed of DNA. DNA carries hereditary information in units called genes. A single chromosome can contain 20,000 or more genes. Genes code for specific proteins that regulate cellular activity and inherited traits (some of which affect carcinogenesis and cancer growth, as well as the efficacy and metabolism of anticancer agents). The genetic information is encoded in DNA by precise sequencing of subunits known as nucleotides. Each nucleotide consists of a sugar (deoxyribose), phosphoric acid, and a base. Four bases exist in DNA: adenine, thymine, guanine, and cytosine. Adenine and guanine are purines, and thymine and cytosine are pyrimidines (Fig. 104-8). These nucleotides are connected linearly to form a chain. Each DNA molecule is made up of two chains of nucleotides, which wind around each other to form a double helix. The two strands are held together by chemical bonding between the bases. The bonding process is very specific—adenine binds only with thymine, and guanine binds only with cytosine. This is known as complementary base pairing. RNA is important in the DNA-directed synthesis of proteins or enzymes. RNA differs from DNA in that it is composed of a single strand of nucleotides, the sugar is ribose, and the base uracil is substituted for thymine. There are three known types of RNA: messenger RNA (mRNA), transfer RNA (tRNA), and ribosomal RNA (rRNA).
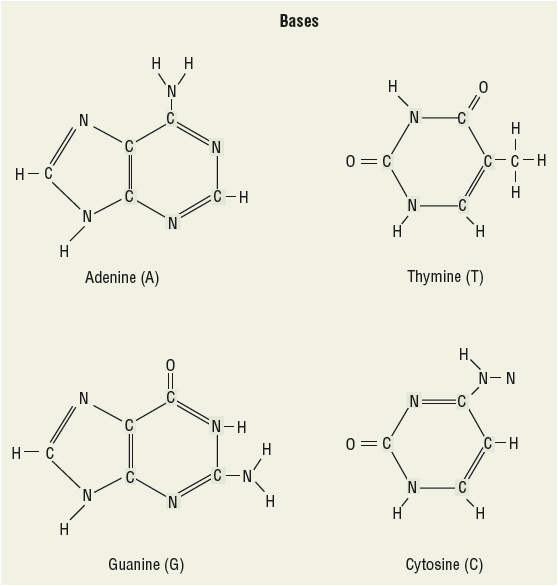
FIGURE 104-8 Structures of DNA bases.
DNA Synthesis
During the DNA synthesis phase (S phase cell cycle), which takes place in the cell nucleus, the DNA unwinds and exposes its nucleotides. When DNA unwinds for replication or protein synthesis, only the portion of the molecule containing the needed nucleotides needs to be exposed. Rather than unwinding the entire strand, topoisomerase I and II enzymes cleave the DNA strands to facilitate unwinding of the section that is needed. The enzyme DNA polymerase matches free complementary nucleotides from the environment to the exposed nucleotides of the DNA. The newly created strands rewind, resulting in two complete double helices. The topoisomerase enzymes are also responsible for resealing the cleaved DNA strands. Most traditional chemotherapies interfere with DNA synthesis.
Protein Synthesis
The synthesis of proteins is a more complex process. Proteins consist of chains of amino acids in very specific sequences. As in DNA synthesis, the double helix must unwind. However, in protein synthesis, only the portion of the DNA molecule that codes for the desired protein is exposed. The enzyme RNA polymerase matches free complementary RNA nucleotides to the exposed DNA nucleotides, and the resultant chain of nucleotides is called mRNA. This process is called transcription. The mRNA travels to ribosomes in the cytoplasm, where protein synthesis occurs. Each three nucleotides of the mRNA chain compose a codon, whose sequence is specific for a particular amino acid. The codon is recognized by tRNA, which then carries the amino acid to the ribosome, where it is added to the growing peptide chain. This process is known as translation. The completed protein is then ready for its intended use as an enzyme or as a structural component. Targeted therapies, such as targeted drugs and MoABs, typically affect protein synthesis, including aberrant growth factor receptors, dysregulated intracellular signaling pathways, and defective apoptosis and angiogenesis. Whereas MoABs affect cell surface receptors, targeted drugs tend to inhibit intracellular signaling. Other anticancer agents that affect proteins include asparaginase and chemotherapy that interfere with microtubules.
CLINICAL PHARMACOLOGY OF ANTICANCER AGENTS
Anticancer agents are commonly categorized by their mechanism of action or by their origin. Akylators exert their effects on DNA and protein synthesis by binding to DNA and preventing the unwinding of the DNA molecule. Antimetabolites resemble naturally occurring nuclear structural components (“metabolites”), such as the nucleotide bases, or inhibit enzymes involved in the synthesis of DNA and proteins. Antitumor antibiotics derive their name from their source; they are fermentation products of Streptomyces species. Figure 104-9 shows the sites of action of common categories of anticancer agents, including traditional chemotherapy, biologic therapies, and targeted drugs. The following sections address these classes of chemotherapies used in the treatment of cancer. The clinical uses, mechanisms, adverse events, and practical patient management for commonly used chemotherapies in each class are detailed. Table 104-8 summarizes dose modifications of individual chemotherapies.
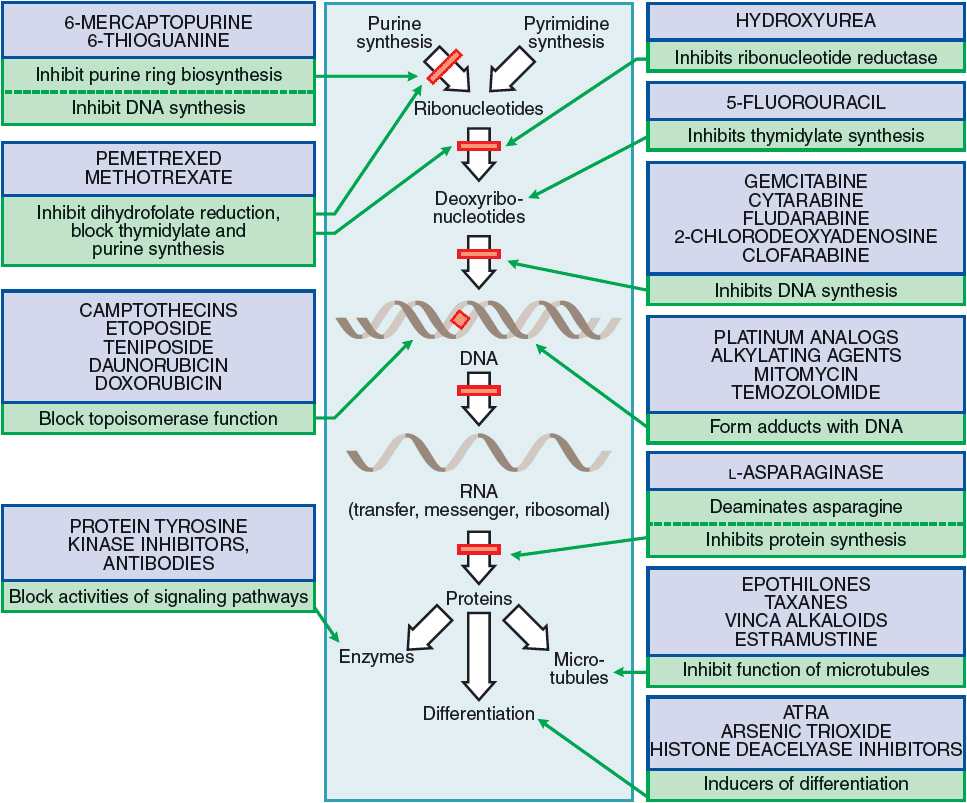
FIGURE 104-9 Mechanisms of action of commonly used anticancer agents. (ATRA, all-trans-retinoic acid.) (From Chabner BA. General Principles of Chemotherapy. In: Brunton LL, Chabner BA, Knollman BC (eds). Goodman & Gilman’s The Pharmacologic Basis of Therapeutics, 12th ed. New York: McGraw-Hill, 2010.)
TABLE 104-8 Monitoring of Anticancer Drugs—a
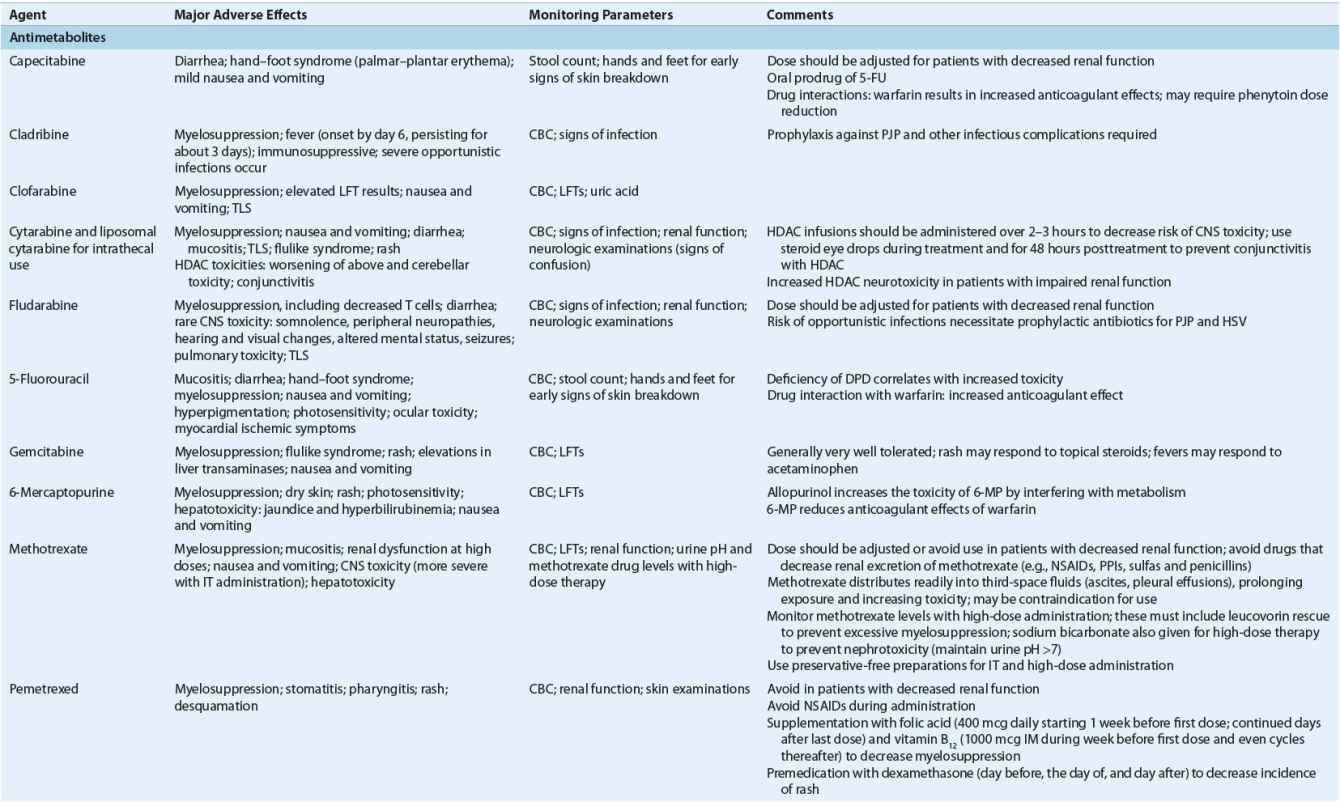
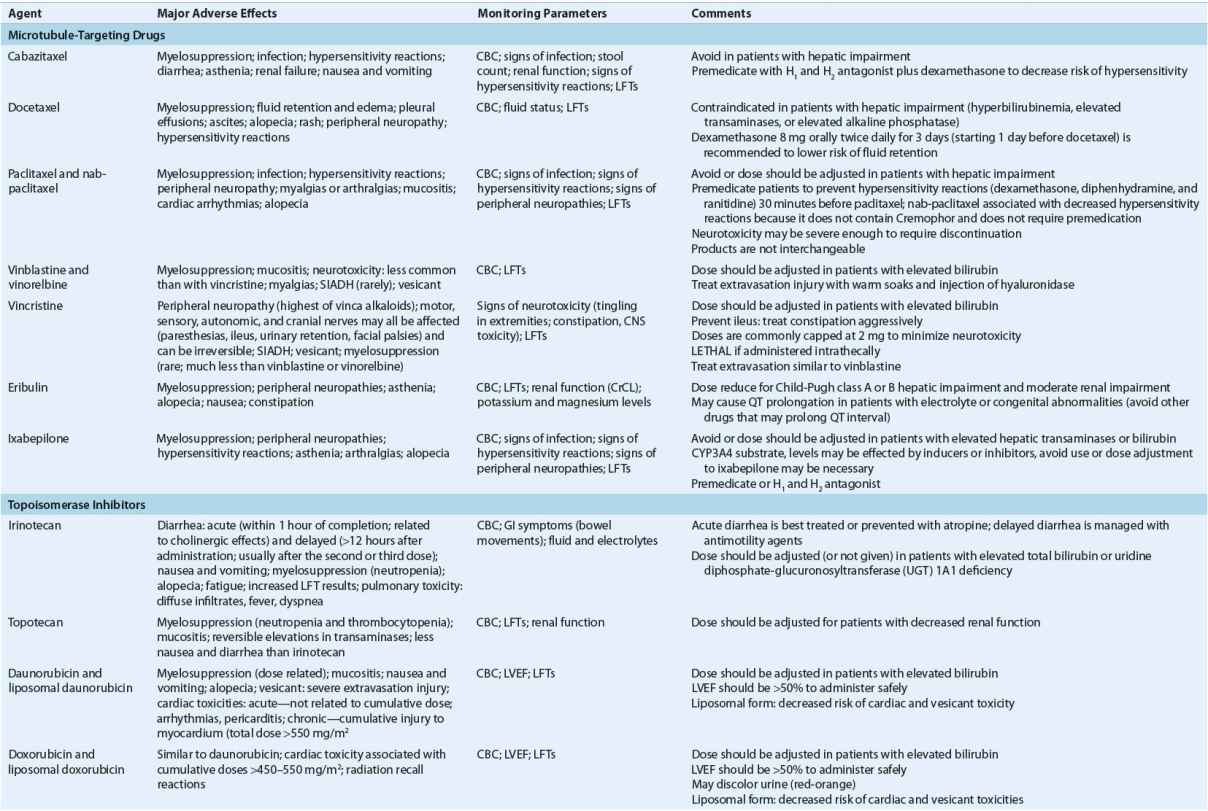
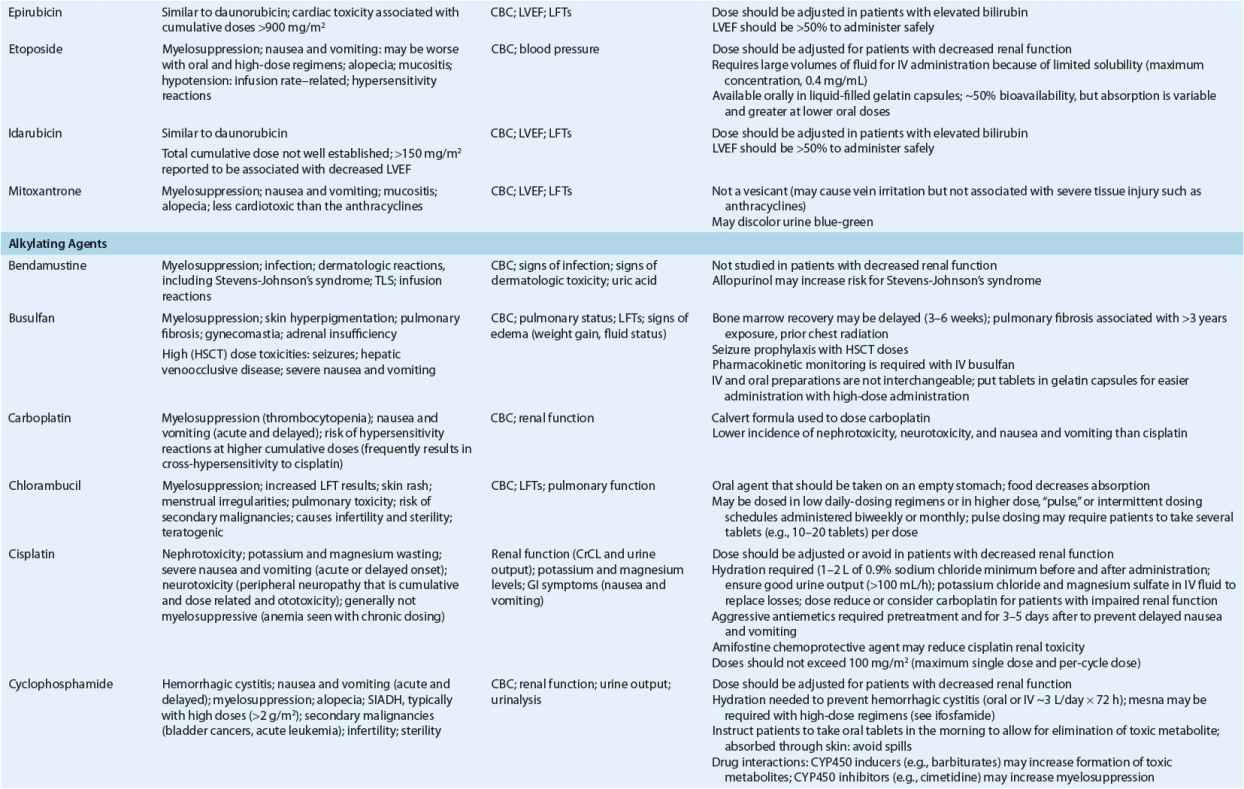
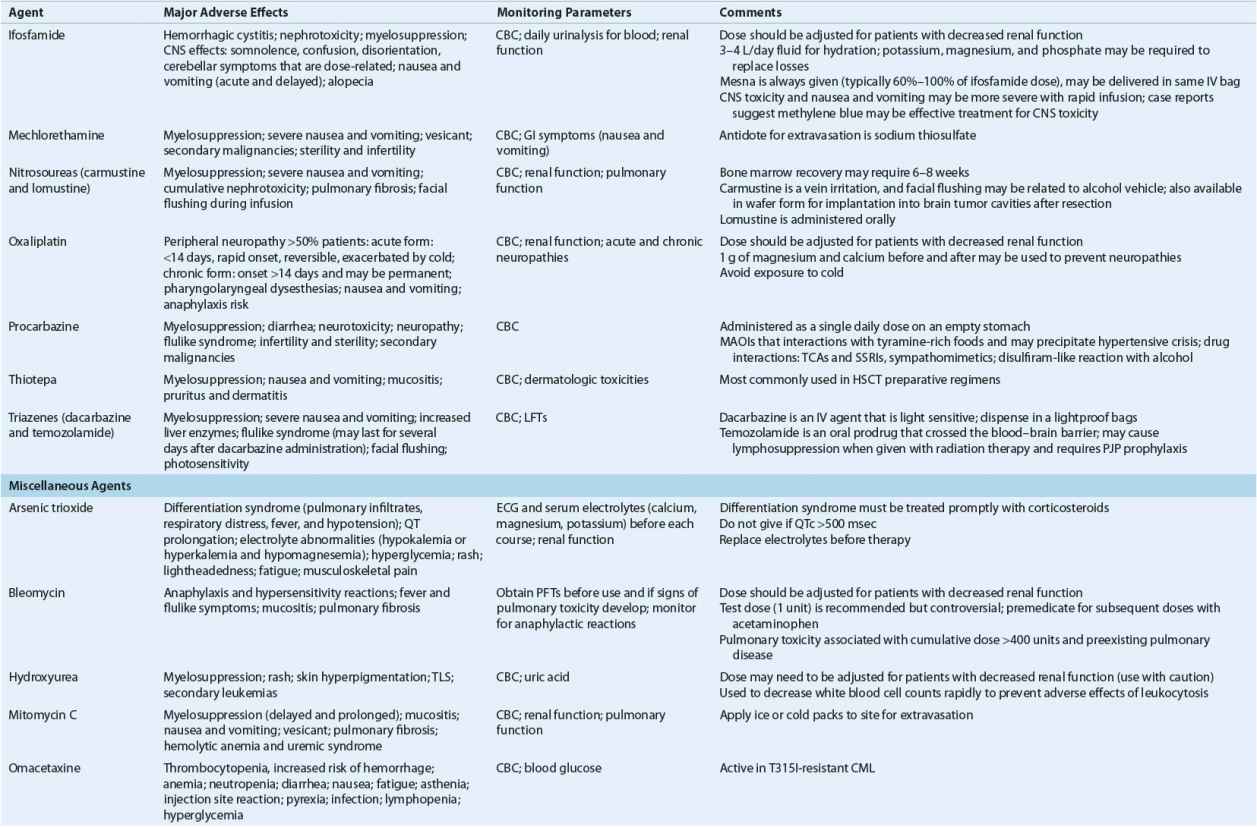
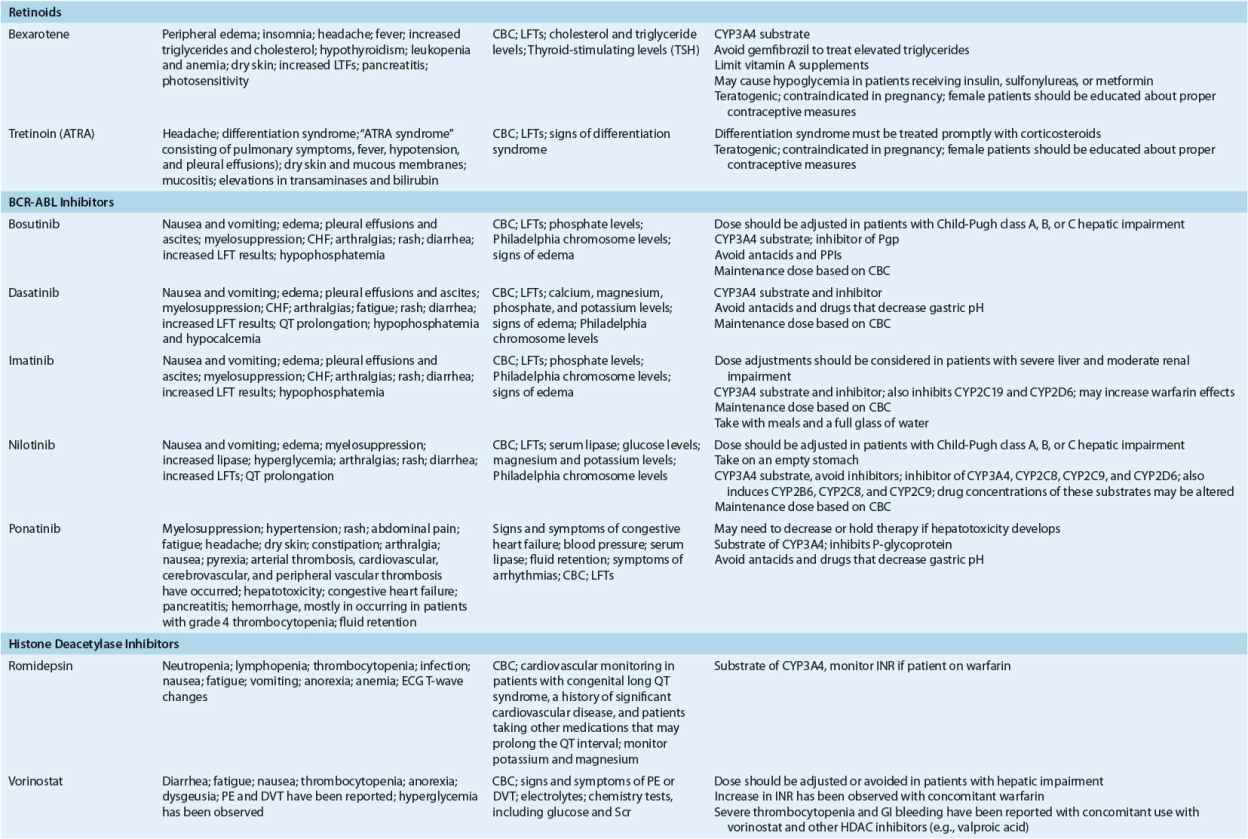
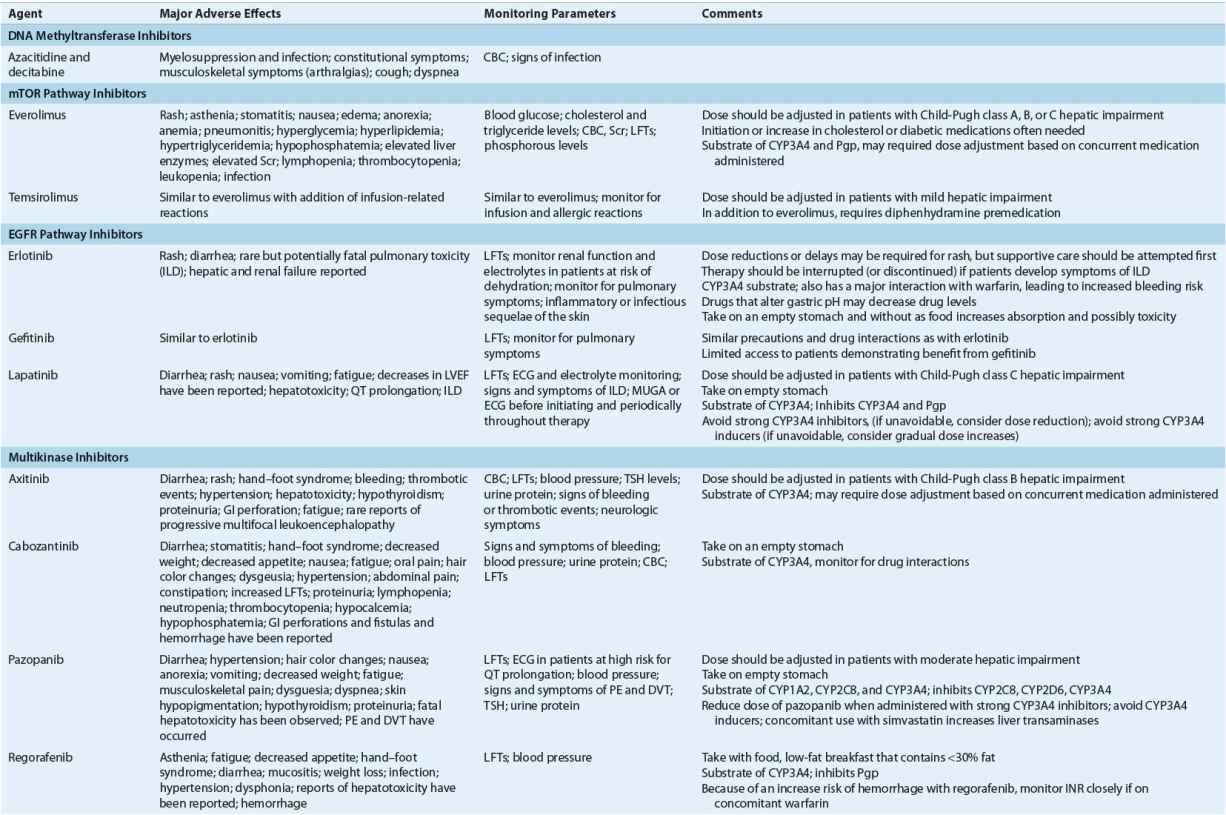
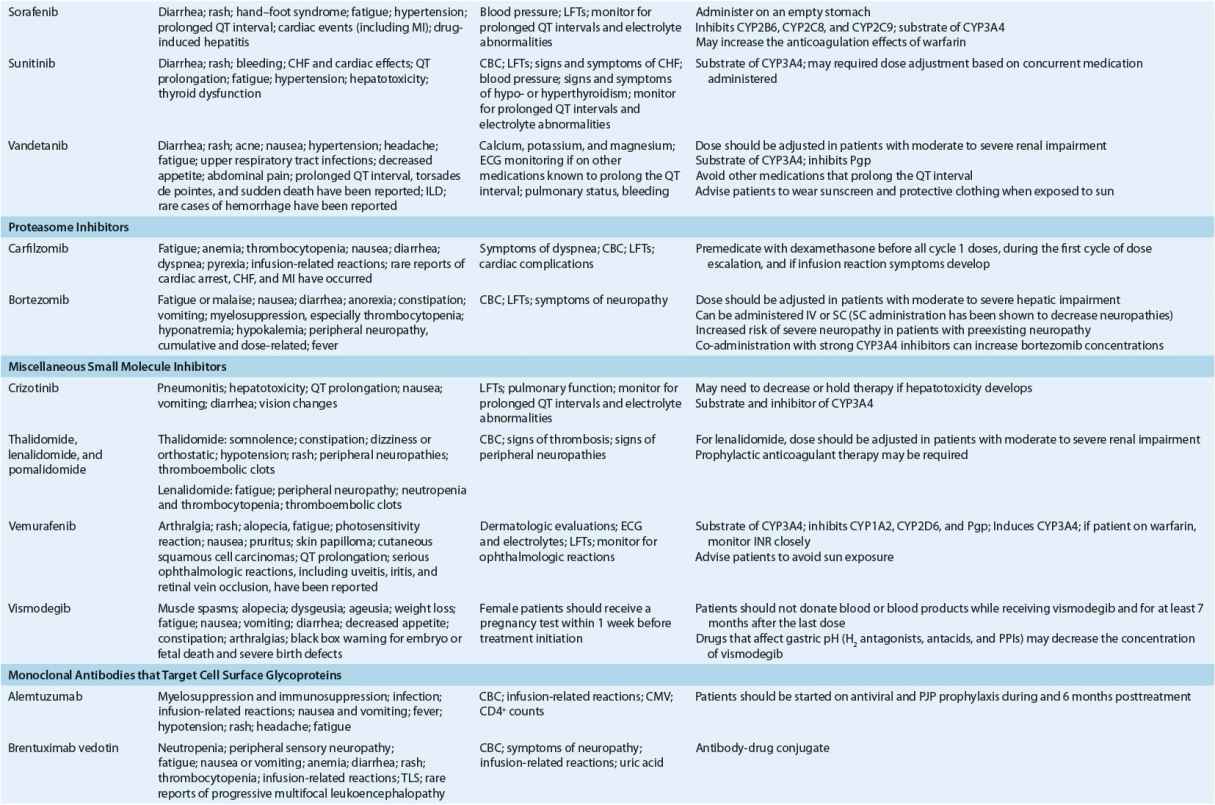
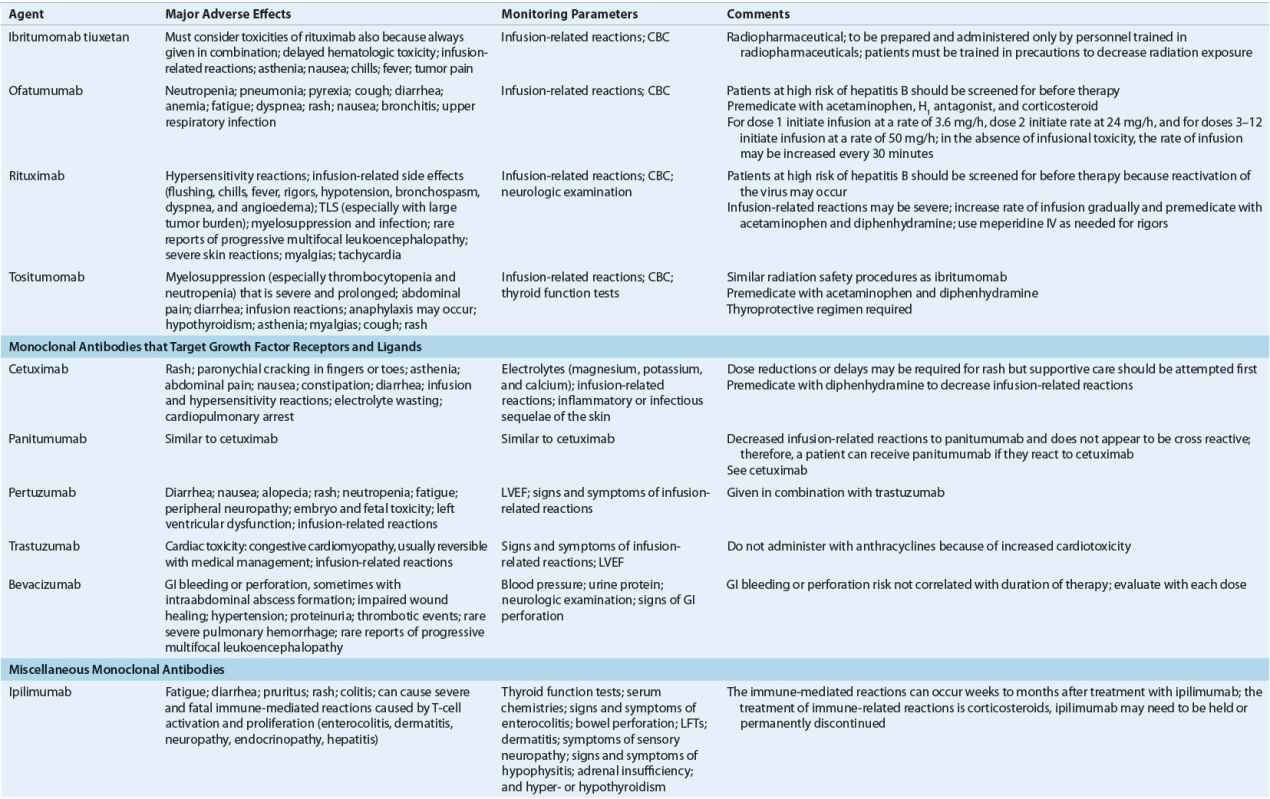
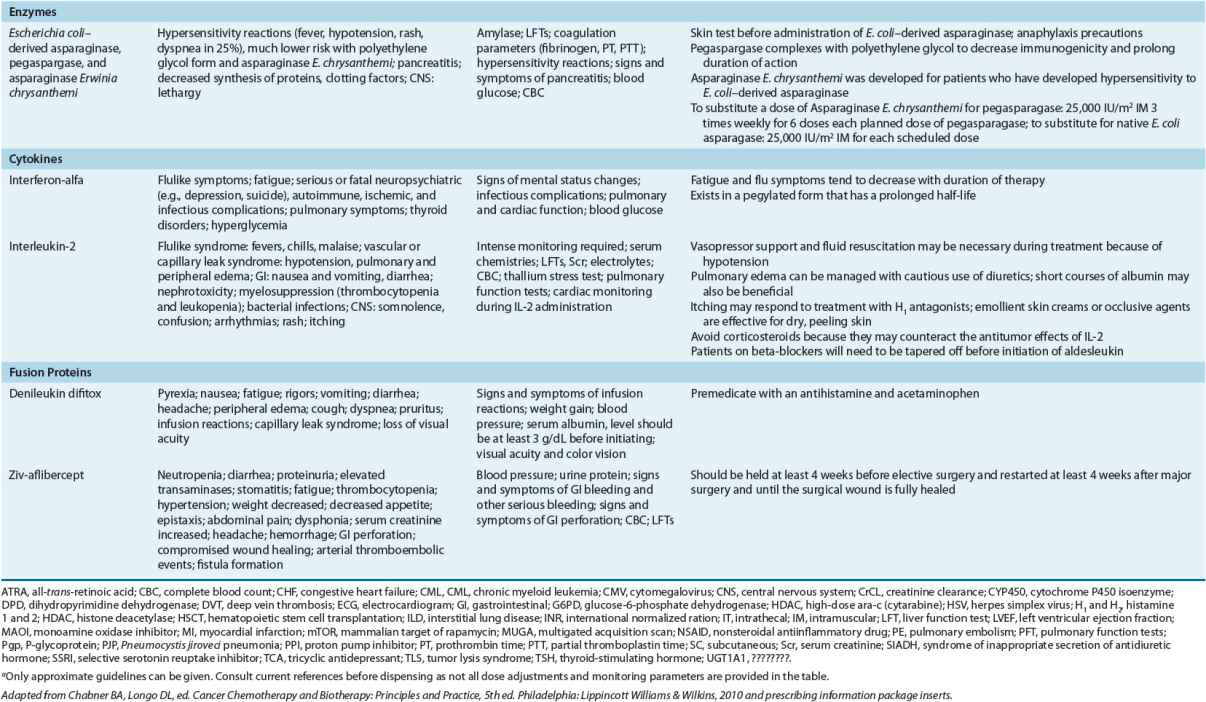
Antimetabolites
Antimetabolites are similar to the nucleotides that make up DNA and RNA. The body mistakes these anticancer agents for the naturally occurring nucleotide bases and metabolizes these agents as the natural nucleotides. These anticancer agents ultimately disrupt replication and cell division by interfering with the production of nucleic acids, DNA, and RNA. Unfortunately, these compounds are not selective for cancer cells, and rapidly dividing normal cells may be poisoned. The most common adverse events associated with the antimetabolites are secondary to a direct cytotoxic effect on rapidly dividing normal cells, such as the bone marrow cells. The three major classes of antimetabolites include pyrimidines, purines, and folate antagonists.
Fluorinated Pyrimidines
5-Fluorouracil 5-Fluorouracil (5-FU) is a fluorinated analog of uracil that was originally synthesized in the late 1950s. It is a prodrug and undergoes sequential phosphorylation to a mono-, di-, and triphsophate similar to natural nucleotide bases to become an active anticancer agent. In the presence of folates, the monophosphate binds tightly to and interferes with the function of thymidylate synthase. This enzyme is required for synthesis of thymidine. The triphosphate metabolite is incorporated into RNA as a false base and interferes with its function. Interference with both thymidine formation and RNA function is important in producing the cytotoxic effects of 5-FU. Although 5-FU nucleotides can also be incorporated directly into DNA and affect its stability, the contribution to cell damage remains unclear. The method of administration influences the mechanism of action, with thymidylate synthesis inhibition playing a greater role in continuous-infusion regimens and incorporation into RNA being more important for intermittent bolus schedules.30
Several pharmacologic strategies have been attempted to increase the cytotoxicity of 5-FU against cancer cells and decrease its toxicity to normal cells. The most common strategy combines 5-FU with the reduced folate leucovorin. Folates increase the stability of the monophosphate-thymidylate synthase complex, thereby increasing the cytotoxicity and clinical activity of 5-FU.30 Dihydropyrimidine dehydrogenase (DPD) catabolize 5-FU and reduced expression of this enzyme has been associated with severe adverse events.31
Capecitabine Capecitabine is an oral pyrimidine uracil analog and is a prodrug of 5-FU. Because capecitabine is enzymatically converted to 5-FU, it shares the same mechanisms of action. It generates higher levels of 5-FU selectively within some tumors as compared with normal tissues. Because chronic twice-daily oral dosing of capecitabine produces sustained 5-FU levels similar to continuous IV infusions of 5-FU, the toxicity pattern is similar to that of a continuous infusion of 5-FU.30,32
The most common toxicities of fluoropyrimidines include neutropenia, thrombocytopenia, and anemia when administered as an IV bolus administration and hand-foot syndrome and diarrhea when administered as a continuous IV infusion.30,32
Cytidine Analogs
Cytarabine Cytarabine (ara-C) is an arabinose analog of cytosine. It is phosphorylated to its active triphosphate within cancer cells and inhibits DNA polymerase, an enzyme responsible for strand elongation. It is also incorporated directly into DNA, where it inhibits the replication of DNA and acts as a chain terminator to prevent DNA elongation. Deaminase enzymes, particularly cytidine deaminase, degrades ara-C.30,33
Cytidine deaminase levels are very low in the central nervous system (CNS). Therefore, cytotoxic concentrations of ara-C are maintained in the CNS for several hours after intrathecal administration of traditional cytarabine formulations and for more than 2 weeks after administration of a depot formulation.
The toxicity of cytarabine is dose dependent. The most characteristic toxicity associated with high-dose ara-C (>1 g/m2 per dose) is a cerebellar syndrome that manifests as dysarthria, nystagmus, and ataxia. The risk of CNS toxicity is strongly correlated with advanced age and renal dysfunction. Renal dysfunction permits accumulation of high levels of the triphosphate, which is believed to be neurotoxic. Hepatic dysfunction, high cumulative doses, and bolus dosing may also increase the risks of neurotoxicity.30,33
Gemcitabine Gemcitabine is a fluorine-substituted deoxycytidine analog that is related structurally to cytarabine. Its activation and mechanism of action are similar to those of cytarabine. Gemcitabine is incorporated into DNA, where it inhibits DNA polymerase activity. It also inhibits ribonucleotide reductase, which is the enzyme required to convert ribonucleotides into the deoxyribonucleotides that are needed for both DNA synthesis and repair. Compared with cytarabine, gemcitabine achieves intracellular concentrations about 20 times higher, secondary to increased penetration of cell membranes and greater affinity for the activating enzyme deoxycytidine kinase. Gemcitabine that is incorporated into DNA has a prolonged intracellular half-life. Its stereoconfiguration causes another normal base pair to be added next to the fraudulent gemcitabine base pair in the DNA strand. This “masked chain termination” protects the gemcitabine from excision and elimination.30,33
Purines and Purine Antimetabolites
Mercaptopurine and Thioguanine 6-Mercaptopurine (6-MP) and its analog thioguanine are rapidly converted to ribonucleotides that inhibit purine biosynthesis. They also undergo purine interconversion reactions needed to supply purine precursors for synthesis of nucleic acids. Clinical cross-resistance is generally observed.30 Both anticancer agents are metabolized by thiopurine methyltransferase (TPMT) and hypoxanthine phosphoribosyl transferase to produce multiple metabolites responsible for the efficacy, hepatic toxicity, and myelosuppression. Genetic polymorphisms of TPMT are associated with reduced enzyme activity and decreased tolerance of standard doses of 6-MP.
6-MP depends on xanthine oxidase for an initial oxidation step. Its metabolism is markedly decreased by concomitant administration of the xanthine oxidase inhibitor allopurinol, and serious toxicity may result. Oral 6-MP doses must be reduced when allopurinol is administered together with 6-MP.30
Fludarabine Monophosphate Fludarabine monophosphate is an analog of the purine adenine. Similar to cytarabine, fludarabine interferes with DNA polymerase, causing chain termination. Fludarabine also incorporates into RNA, resulting in inhibited transcription. The usual dose-limiting toxicity is myelosuppression. Fludarabine is also immunosuppressive, with associated opportunistic infections resulting from its effect on T cells and a subsequent decrease in CD4 counts; prophylactic antibiotics and antiviral medications are recommended and should continue until CD4 counts normalize.30,34
Cladribine and Pentostatin Cladribine and pentostatin are purine nucleoside analogs with slightly different mechanisms of action. Cladribine is resistant to inactivation by adenosine deaminase and triphosphorylated to an active form that is incorporated into DNA, resulting in inhibition of DNA synthesis and early chain termination. Its antitumor activity is unusual for an antimetabolite in that it affects both actively dividing and resting cancer cells. Pentostatin is a potent inhibitor of adenosine deaminase. Adenosine deaminase is an enzyme critical in purine base metabolism and is found in high concentrations in lymphatic tissue. Similar to fludarabine, these chemotherapies have immunosuppressive effects that place patients at risk for serious opportunistic infections.30,33
Antifolates
Folate vitamins are essential cofactors in DNA synthesis. These vitamins carry one-carbon groups in transfer reactions that are required for purine and thymidylic acid synthesis. Natural folates circulating in the blood have a single glutamic acid group, but natural folates within cells are converted to polyglutamates, which are more efficient cofactors and preferentially retained inside cells.35
Dietary folates must be chemically reduced to their tetrahydrofolate forms to be active. The enzyme responsible for this reduction is dihydrofolate reductase (DHFR). Antifolates are associated with neutropenia and thrombocytopenia, mucositis, and nausea and vomiting.
Methotrexate Methotrexate (MTX) inhibits DHFR, which results in the depletion of intracellular pools of reduced folates (tetrahydrofolates) essential for thymidylate and purine synthesis. Lack of either thymidine or purines prevents synthesis of DNA. The DHFR-mediated effects of antifolates on normal and tumor cells may be neutralized by supplying reduced folates exogenously. The reduced folate used clinically for “rescue” is leucovorin (folinic acid), which bypasses the metabolic block induced by DHFR inhibitors.35
Amplification of DHFR can lead to tumor cell resistance. Other potential causes of resistance are slow rates of thymidylate synthesis, decreased affinity for DHFR, lack of polyglutamation within cancer cells and saturated transport. In high doses, passive diffusion may overcome tumor cell resistance caused by saturated active transport systems. Malignant cells may achieve greater MTX polyglutamate levels than normal cells, which may, in part, explain the selective effects of MTX on malignant versus normal cells.35
Accurate and readily available assays for serum MTX levels have made therapeutic drug monitoring of MTX a valuable clinical tool. The threshold for cytotoxic effects of MTX is about 0.02 mg/L (50 nmol/L). Toxicity and efficacy relates not only to peak concentrations, but more importantly to time that concentrations remain above this threshold level. For MTX doses requiring leucovorin rescue (generally doses greater than 1,000 mg/m2), leucovorin must be administered until levels fall below 0.02 mg/L (50 nmol/L). Therapeutic drug monitoring is also an effective means of increasing the likelihood of therapeutic success by individualizing doses based on target levels.35 Renal tubular necrosis is seen with high-doses of MTX and vigorous hydration with or without alkalinization of the urine necessary to decrease risk of renal failure.
Glucarpidase has been approved for the treatment of toxic plasma MTX concentrations in patients with delayed MTX clearance because of impaired renal function. It is important to note that MTX concentrations within 48 hours after glucarpidase administration can only be reliably measured by chromatographic methods. Immunoassays can overestimate MTX concentration because of interference from metabolites.36
Pralatrexate Pralatrexate is an antifolate drug approved for patients with relapsed or refractory peripheral T-cell leukemias. It competitively inhibits DHFR. It is also a competitive inhibitor for polyglutamylation by the enzyme folylpolyglutamyl synthetase. This inhibition results in the depletion of thymidine and other synthesis of biological molecules that depends on single carbon transfer.37
Pemetrexed Pemetrexed is a multitargeted antifolate that inhibits at least three biosynthetic pathways in thymidine and purine synthesis. In addition to inhibiting DHFR, it also inhibits thymidine synthase and glycinamide ribonucleotide formyltransferase, decreasing the risk of the development of drug resistance. Severe hematologic toxicity and deaths associated with neutropenic sepsis have been reported in clinical trials. Elevated baseline cystathionine or homocysteine concentrations correlated with this unexpected toxicity. Routine supplementation of folic acid and vitamin B12 lowers levels of these substances and lowers the risk of mortality related to neutropenic sepsis. The approved labeling of pemetrexed requires administration of folic acid and vitamin B12 throughout the duration of treatment.27
Microtubule-Targeting Drugs
Vinca Alkaloids
Vincristine, vinblastine, and vinorelbine are natural alkaloids derived from the periwinkle (vinca) plant. They act as mitotic inhibitors, or “spindle poisons.” Although the alkaloids are very similar structurally, they have different activities and patterns of toxicity. Whereas vinorelbine and vinblastine are associated with dose-limiting myelosuppression, vincristine causes mild myelosuppressive effects but is more neurotoxic.
Vinca alkaloids bind to tubulin, the structural protein that polymerizes to form microtubules. These hollow tubes make up the mitotic spindle and are important in nerve conduction and neurotransmission. Vinca alkaloids disrupt the normal balance between polymerization and depolymerization of microtubules, inhibiting assembly of microtubules and disrupting microtubule dynamics. This interferes with formation of the mitotic spindle and causes cells to accumulate in mitosis. They also disturb a variety of microtubule-related processes in cells and induce apoptosis. Resistance to the vinca alkaloids develops primarily from P-glycoprotein (Pgp)-mediated multidrug resistance, which decreases drug accumulation and retention within cancer cells.38
Taxanes
Paclitaxel and docetaxel are taxane plant alkaloids with antimitotic activity. Paclitaxel was isolated from the bark of the Pacific yew tree, Taxus brevifolia, but is now produced semisynthetically from the needles of the European yew, Taxus baccata. Docetaxel is a semisynthetic taxoid extracted from 10-deacetyl baccatin III, a noncytotoxic precursor found in the renewable needle biomass of yew plants.38
Paclitaxel and docetaxel both act by binding to tubulin, but unlike the vinca alkaloids, they do not interfere with tubulin assembly. Instead, the taxanes promote microtubule assembly and therefore interfere with microtubule disassembly. They induce tubulin polymerization, resulting in formation of inappropriately stable, nonfunctional microtubules. The stability of the microtubules damages cells by disrupting the dynamics of microtubule-dependent structures required for mitosis and other cellular functions. Taxanes also have some nonmitotic actions that can promote cancer cell death, such as inhibition of angiogenesis. Resistance to the antitumor effects of the taxanes is attributable to alterations in tubulin or tubulin binding sites or to Pgp multidrug resistance. Although paclitaxel and docetaxel have very similar mechanisms of action, cross-resistance between the two chemotherapies is incomplete.38 Myelosuppression is common with both taxanes, but other adverse events can differ. While increased fluid retention is seen with docetaxel, increased neurotoxicity and hypersensitivity reactions are seen with paclitaxel.27,38 Both require premedications with corticosteroids; paclitaxel also requires antihistamines to decrease the likelihood of hypersensitivity reactions.
To circumvent the hypersensitivity reactions with paclitaxel and to possibly increase its efficacy, paclitaxel was formulated to be bound to albumin (nab-paclitaxel). This new dosage form is devoid of the Cremophor excipient that is believed to mediate the hypersensitivity reactions and exacerbate myelosuppression with the conventional formulation. This formulation appears to be selectively activated by cancer cells to the active paclitaxel compound.39 In clinical trials, nab-paclitaxel has shown comparable activity to the conventional formulation of paclitaxel with a lower incidence of hypersensitivity reactions. Peripheral neuropathies remain a common adverse event with this formulation.
Cabazitaxel is a new semisynthetic derivative of docetaxel that has been demonstrated to elicit an antitumor response in tumors resistant to paclitaxel and docetaxel despite having the same mechanism of action. This is partially because of its lack of affinity for Pgp mentioned earlier that allows cabazitaxel to remain inside the cancer cells. Adverse events and premedications are similar to traditional taxanes.40
Epothilones
Similar to the taxanes, the epothilones work in the M phase of the cell cycle. Epothilone binding to microtubules is distinct from taxanes with activity demonstrated in paclitaxel-resistant cell lines.41 Epothilones appear to be poor substrates for Pgp and their cytotoxicity is not affected by its overexpression. Natural epothilones are macrolide derivatives that have stability and pharmacokinetic problems. Synthetic anticancer agents have been developed with the epothilone, ixabepilone, approved for the treatment of metastatic breast cancer. Toxicities are similar to those of the taxanes; premedication with antihistamines are required, although no corticosteroid is administered unless the patient experiences a hypersensitivity reaction to a previous dose.
Halichondrins
Eribulin is a nontaxane antimicrotuble analogue of the macrolide halichondrin B. It was originally isolated from the marine sponge Halichondria okadai but is now fully synthetic. Similar to the vinca alkaloids, eribulin inhibits tubulin polymerization by inhibiting microtubule growth; however, in contrast, it does not shorten or promote depolymerization of microtubules.42 Additionally, eribulin only binds to the β-tubulin subunit and has demonstrated the ability to overcome taxane resistance conferred by β-tubulin mutations.42 Adverse events are similar to those of vinblastine (e.g., neutropenia), with a decreased incidence of neuropathy compared with vincristine and taxanes.
Estramustine
Estramustine is an unusual drug, because it structurally combines the alkylating agent nor-nitrogen mustard with estradiol.27 It was designed with the intent that the estradiol portion of the molecule would facilitate uptake of the alkylating agent into hormone-sensitive prostate cancer cells. Despite the inclusion of an alkylator, estramustine does not function in vivo as an alkylating agent. The estradiol is released after its administration and is responsible for most of the toxicity associated with estramustine, but it is not believed to contribute to its cytotoxic effect. In the mid 1980s, estramustine was redefined as an antimicrotubule agent. It binds covalently to microtubule-associated proteins that are part of the structural support for microtubules. The binding causes the separation of microtubule-associated proteins from the microtubules, inhibiting microtubule assembly and eventually causing their disassembly.27
Topoisomerase Inhibitors
Topoisomerases are essential enzymes involved in maintaining DNA topologic structure during replication and transcription. DNA topoisomerase enzymes relieve torsional strain during DNA unwinding by producing strand breaks. They cleave DNA strands and form intermediates with the strands, producing a gap through which DNA strands can pass, and then reseal the strand breaks. Topoisomerase I produces single-strand breaks; topoisomerase II produces double-strand breaks.43 Several important anticancer agents interact with topoisomerase enzymes: camptothecins, anthracyclines, and the epipodophyllotoxins.
Camptothecin Derivatives
The camptothecin analogs irinotecan and topotecan were synthesized to reduce toxicity and improve therapeutic effects of camptothecin, a plant alkaloid derived from Camptotheca acuminata. Topotecan and irinotecan, through its active metabolite SN-38, inhibit topoisomerase I enzyme activity. Topoisomerase I enzymes stabilize DNA single-strand breaks and inhibit strand resealing.43,44 Irinotecan undergoes metabolism to SN-38 by the polymorphic enzyme uridine diphosphate glucosyltransferase, and variant tandem repeats in the promoter of this gene are associated with a higher risk of diarrhea and neutropenia.
Etoposide and Teniposide
Etoposide and teniposide are semisynthetic podophyllotoxin derivatives that bind to tubulin and interfere with microtubule formation. Etoposide and teniposide also damage cancer cells by causing strand breakage through inhibition of topoisomerase II.43 Resistance may be caused by differences in topoisomerase II levels, increased cell ability to repair strand breaks, or increased levels of Pgp. Etoposide and teniposide are usually clinically cross-resistant. They are cell-cycle phase specific and arrest cells in the S or early G2 phase. As a result, activity is much greater when they are administered in divided doses over several days rather than in large single doses.
Anthracene Derivatives
The most widely used and best understood anthracene derivative is doxorubicin (Adriamycin or “Adria”). Other members of the anthracene group include daunorubicin (daunomycin), idarubicin, epirubicin, and mitoxantrone. All of these derivatives, except mitoxantrone, are anthracyclines and share a common, four-membered anthracene ring complex with an attached aglycone or sugar portion. The ring complex is a chromophore and accounts for the intense colors of these derivatives.43,45
Doxorubicin, Daunorubicin, Idarubicin, and Epirubicin Anthracyclines are classified as antitumor antibiotics, but they have multiple mechanisms of action. Although anthracyclines can intercalate into DNA and cause structural changes that interfere with DNA and RNA synthesis, this is not their primary mechanism of cytotoxicity. Intercalating anticancer agents insert or stack between base pairs of DNA. However, anthracyclines primarily inhibit topoisomerase II, producing double-strand DNA breaks.27,43,45
The anthracyclines also undergo electron reductions to reactive compounds that can damage DNA and cell membranes. Free radicals formed from reduction of the anthracyclines first donate electrons to oxygen to make superoxide, which can react with itself to make hydrogen peroxide. Cleavage of hydrogen peroxide produces the highly reactive and destructive hydroxyl radical. This last step requires iron, and anthracyclines are potent iron binders. Iron-anthracycline complexes can then bind to DNA and react rapidly with hydrogen peroxide to produce the hydroxyl radicals that actually cleave DNA. Human cells have natural defenses against oxygen radical damage, in the form of enzymes that can convert the radicals to less reactive compounds, or that can repair DNA damage. Differences in distribution of these defensive enzymes may account for the cumulative dose limiting cardiotoxicity associated with anthracyclines. For example, cardiac muscle has low levels of defensive enzymes and high levels of enzymes that activate anthracyclines. Oxygen free-radical formation is firmly established as a cause of cardiac damage and extravasation injury but is not a major mechanism of tumor-cell killing. Resistance to anthracyclines is usually secondary to Pgp-dependent multidrug resistance. Altered topoisomerase II activity may contribute to the development of resistance.27,45
Mitoxantrone Mitoxantrone was synthesized in an attempt to develop a chemotherapy with comparable antitumor activity to doxorubicin but with an improved safety profile. Similar to the anthracyclines, mitoxantrone is an intercalating topoisomerase II inhibitor, but its potential for free-radical formation is much less than that of the anthracyclines. This decreased tendency for free-radical formation may explain the reduced risks of cardiac toxicity and ulceration after extravasation.27,45
Alkylating Agents
The alkylating agents are among the oldest and most useful classes of anticancer agents. Their clinical use evolved from the observation of bone marrow suppression and lymph node shrinkage in soldiers exposed to sulfur mustard gas warfare during World War I.27 In an effort to develop similar agents that might be useful in treating cancerous overgrowths of lymphoid tissues, less reactive derivatives were synthesized. Their effectiveness as anticancer agents was confirmed by clinical trials in the mid-1940s.
All alkylating agents work by covalently bonding to highly reactive alkyl groups or substituted alkyl groups with nucleophilic groups of proteins and nucleic acids. Some agents react directly with biologic molecules, but others form an intermediate compound that reacts with these molecules. The most common binding site for alkylating agents is the seven-nitrogen group of the DNA base guanine. These covalent interactions result in cross-linking between two DNA strands or between two bases in the same strand of DNA. Reactions between DNA and RNA and between drug and proteins may also occur, but the main insult that results in cell death is inhibition of DNA replication because the interlinked strands do not separate as required. Because the alkylating agents can damage DNA during any phase of the cell cycle, they are not cell-cycle phase specific. However, their greatest effect is seen in rapidly dividing cells.
As a class, alkylators are cytotoxic, mutagenic, teratogenic, carcinogenic, and myelosuppressive. Resistance to these chemotherapies can occur from increased DNA repair capabilities, decreased entry into or accelerated exit from cells, increased inactivation inside cells, or lack of cellular mechanisms to result in cell death after DNA damage. They react with water and are inactivated by hydrolysis, making spontaneous degradation an important component of their elimination.46
Nitrogen Mustards
Cyclophosphamide and Ifosfamide Cyclophosphamide and ifosfamide are nitrogen mustard derivatives and are widely used in the treatment of solid tumors and hematologic malignancies. These mustards are closely related in structure, clinical use, and toxicity. Neither agent is active in its parent form and must be activated by cytochrome P450 (CYP) enzymes. One of the active metabolites of cyclophosphamide is phosphoramide mustard and of ifosfamide is ifosfamide mustard. The CYP-mediated metabolites 4-hydroxy-cyclophosphamide and 4-hydroxyifosfamide are also cytotoxic compounds. Acrolein, a metabolite of both cyclophosphamide and ifosfamide, has little antitumor activity, but is responsible for the hemorrhagic cystitis associated with ifosfamide and sometimes high-dose cyclophosphamide.46 Encephalopathy after ifosfamide can occur within 48 to 72 hours after the infusion and is reversible. The increased production of dechloroethylated metabolites after administration of ifosfamide compared with cyclophosphamide may explain the increased risk of CNS toxicity associated with ifosfamide.47
Bendamustine Bendamustine is an alkylating agent (nitrogen mustard derivative) with a benzimidazole ring (purine analog) that demonstrates only partial cross-resistance (in vitro) with other alkylating agents.48 It leads to cell death via single- and double-strand DNA cross-linking, and it is active against quiescent and dividing cells. The primary cytotoxic activity is due to bendamustine rather than its metabolites. It is used primarily to treat lymphoid malignancies, such as chronic lymphocytic leukemia (CLL) and NHL.
Nitrosoureas
The nitrosoureas are alkylating agents characterized by lipophilicity and ability to cross the blood-brain barrier. Carmustine or bischloroethylnitrosourea (BCNU) and lomustine (CCNU) are commercially available. BCNU is available as an IV preparation and as a drug-impregnated biodegradable wafer (Gliadel) for direct application to residual tumor tissue after surgical resection of brain tumors. The nitrosoureas decompose to reactive alkylating metabolites and to isocyanate compounds that have several effects on reproducing cells.46
Nonclassic Alkylating Agents
Several other cytotoxic chemotherapies appear to act as alkylators, although their structures do not include the classic alkylating groups. They are capable of binding covalently to cellular components and include procarbazine, dacarbazine, temozolomide, and the heavy metal compounds.46
Dacarbazine and Temozolomide Dacarbazine and temozolomide are nonclassic alkylating agents. Both compounds undergo demethylation to the same active intermediate (monomethyl triazeno-imidazole-carboxamide [MTIC]) that interrupts DNA replication by causing methylation of guanine. Unlike dacarbazine, temozolomide does not require the liver for activation and is chemically degraded to MTIC at physiologic pH. Both agents inhibit DNA, RNA, and protein synthesis.27,46
Important pharmacokinetic differences exist between these two agents. Dacarbazine is poorly absorbed and must be administered by IV infusion. Temozolomide is rapidly absorbed after oral administration and is nearly 100% bioavailable when given on a completely empty stomach. Dacarbazine penetrates the CNS poorly, but temozolomide readily crosses the blood-brain barrier, achieving therapeutically active concentrations in cerebrospinal fluid and brain tumor tissues.27,46
Cisplatin, Carboplatin, and Oxaliplatin The platinum derivatives—cisplatin, carboplatin, and oxaliplatin—are anticancer agents with remarkable usefulness in cancer treatment. Recognition of cisplatin’s cytotoxic activity was the result of a serendipitous observation that bacterial growth in culture was altered when an electric current was delivered to the media through platinum electrodes. The growth change was noted to be similar to that produced by alkylating agents and radiation. It was found that a platinum-chloride complex, now known as cisplatin, generated by the current was responsible for the changes. Carboplatin is a structural analog of cisplatin in which the chloride groups of the parent compound are replaced by a carboxycyclobutane moiety. It shares a similar spectrum of clinical activity with cisplatin, and cross-resistance is common. Oxaliplatin is an organoplatinum compound in which the platinum is complexed with an oxalate ligand as the leaving group and to diaminocyclohexane. Its spectrum of activity differs substantially from the other platinum compounds and includes notable activity against colorectal cancers.27,46
The cytotoxicity of the platinum derivatives depends on platinum binding to DNA and the formation of intrastrand cross-links or adducts between neighboring guanines. These intrastrand links cause a major bending of the DNA. They may cause cellular damage by distorting the normal DNA conformation and preventing bases that are normally paired from lining up with each other. Interstrand cross-links also occur.27,46
The cytotoxic form of cisplatin is the aquated species in which hydroxyl groups or water molecules replace the two chloride groups. This reaction occurs readily in low concentrations of chloride, such as the concentrations present within cells, and produces a positively charged compound that can react with DNA. The aquated species is responsible for both the efficacy and toxicity of cisplatin. Carboplatin also undergoes aquation but at a slower rate. Oxaliplatin becomes active when the oxalate ligand is displaced in physiologic solutions.27,46
Resistance to the therapeutic effects of platinum compounds may occur through several mechanisms. The ability to repair platinum-induced DNA damage may be increased, or the compounds may be inactivated by increased levels of intracellular glutathione, metallothioneins, or other thiol-containing proteins. Altered uptake into cells may also affect sensitivity to platinum compounds.27,46
Cisplatin is a highly toxic anticancer agent that can cause serious nephrotoxicity, ototoxicity, peripheral neuropathy, emesis, and anemia. The significant efficacy of cisplatin against many tumors makes it a valuable agent despite these toxicities, most of which can be prevented or managed with aggressive supportive care measures.27 In contrast, carboplatin administration is limited by hematologic toxicity. Patients with compromised renal function require dose reductions to limit myelosuppressive toxicity.27,46 The most widely used dosage schema, the Calvert formula (Table 104-9), uses a target area under the curve and renal function parameters to estimate the carboplatin dose. Carboplatin’s potential to cause renal damage, peripheral neuropathy, ototoxicity, and nausea and vomiting is much less than that of comparable cisplatin doses.46 Oxaliplatin is not nephrotoxic or ototoxic and is moderately emetogenic, but it can cause peripheral neuropathies and unique cold-induced neuropathies.49 All of the platinum derivatives have potential to cause hypersensitivity reactions, including anaphylaxis, after a threshold exposure is reached.
TABLE 104-9 Dosing Formulas for Chemotherapy




