FAMILIAL NON-MARFAN DISSECTIONS
Familial aortic dissections are about as common in patients without extravascular abnormalities as in patients with Marfan syndrome (3). The genetic basis is not known, although it has been shown that 5% of patients with non-Marfan familial dissection have mutations in the TGF-β receptor, as is the case of the recently described Loeys-Dietz syndrome (4). As with Marfan syndrome, the histologic characteristic is cystic medial degeneration, although the degree is quite variable.
EHLERS-DANLOS SYNDROME
Ehlers-Danlos syndrome type IV, the vascular type, results from mutations in the gene for type III procollagen (COL3A1). Affected patients are at risk for arterial, bowel, and uterine rupture (5). The histologic features of Ehlers-Danlos syndrome are not well characterized; indeed, in many cases, the media is histologically normal, and the pathologic features are that of rupture and pseudoaneurysm formation in an otherwise apparently normal vessel. In the Mayo Clinic series of aortic root resection, primarily for aneurysm, only 2 of 513 patients had evidence of Ehlers Danlos syndrome, less than 5% of hereditary cases. It is unclear whether cystic medial degeneration is a typical finding of the disease, in contrast to Marfan syndrome and other non-Marfan familial dissections.
AORTIC ROOT DISEASE IN PATIENTS WITH BICUSPID AORTIC VALVE
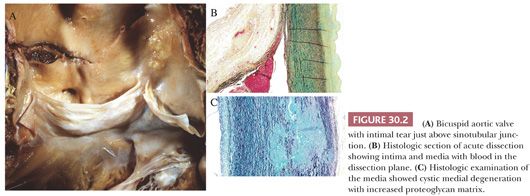
Bicuspid aortic valve (or congenitally bicommissural valve) has an autosomal dominant inheritance (6). There is a known association with aortic root aneurysm and dissection, the risk being 5 to 10 times that of the general population (7). In the Mayo Clinic series of surgically resected ascending aorta, more than 10% of patients (67 of 513) had a bicuspid aortic valve; the incidence in the normal population is less than 2%. In this series, the mean age was 53 years, and the degree of cystic medial necrosis was significantly less in patients with congenitally bicuspid aortic valve as compared to other inherited connective tissue disease; only 11% had severe cystic medial degeneration (3) (Fig. 30.2). However, it should be emphasized that in an individual case, histologic findings of the aortic media cannot distinguish the underlying etiology of medial degeneration. There is genetic evidence that there is a common gene that underlies bicuspid aortic valve and aortic aneurysm and that the most dilated portion of the aorta is distal to the annulus (6). The genetic basis remains unknown, however, and is likely unrelated to fibrillin-1 because patients with Marfan syndrome have trileaflet aortic valves.
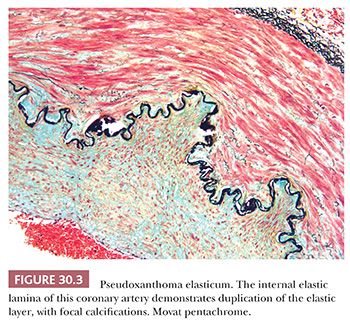
PSEUDOXANTHOMA ELASTICUM
Pseudoxanthoma elasticum (PXE) is caused by mutations in the adenosine triphosphate (ATP)–binding cassette transporter C6 (ABCC6), also known as multidrug resistance–associated protein 6 (MRP6) gene. It is inherited as an autosomal recessive trait and has an incidence of 1 in 25,000 to 100,000. PXE is characterized by progressive calcification and fragmentation of elastic fibers in the skin, the retina, and the cardiovascular system (Fig. 30.3). Patients typically have a normal life span, the morbidity varying based on the extent of extracutaneous involvement.
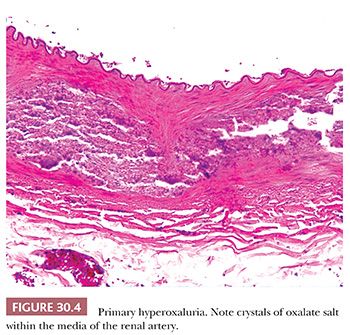
PRIMARILY HYPEROXALURIA
Type I hyperoxaluria occurs in 1 per 120,000 live births and is transmitted as an autosomal recessive trait. It is caused by a deficiency of the peroxisomal liver-specific alanine: glyoxylate aminotransferase gene (i.e., AGT). Most patients develop renal failure in childhood. Occasionally, extensive crystalline deposits within the walls of blood vessels (Fig. 30.4) may result in vasculopathy, clinically mimicking systemic vasculitis.
OTHER NONINFLAMMATORY VASCULAR DISEASES
ACQUIRED AORTIC ROOT DILATATION AND DISSECTION
The majority of patients with degenerative aortic medial disease have no family history or evidence of inherited connective tissue disease. Many of the patients, who are on average 10 to 20 years older than patients with inherited diseases and bicuspid aortic valve, have systemic hypertension. In approximately 25% of patients, however, there are no known predisposing factors. It is likely that these patients have a polygenetic predisposition to aortic dissection. The histologic features of idiopathic aortic root aneurysm, with or without dissection, are similar to those of inherited disease, although the degree of cystic medial degeneration is typically mild. Again, the histologic features per se do not point to an underlying etiology.
GENERAL APPROACH TO ASCENDING AORTIC ANEURYSM
The surgical pathologist, when evaluating an aortic root aneurysm, should recognize the limitations of histologic assessment in assigning an etiology (Table 30.1). Inflammatory lesions, such as idiopathic aortitis, should be excluded because there may be foci mimicking cystic medial necrosis in the otherwise inflamed aortic media. In noninflammatory aortic aneurysms, the degree of medial degeneration is quite variable and does not point to a specific etiology. The number of aortic valve leaflets should be noted in cases with concomitant valve replacement, and the presence of acute dissection should be noted. In addition, any areas of healed dissection, characterized by elastic reduplication of the false lumen lining, should be described; these are readily detected by the use of elastic stains. Adequate sampling of the lesions (approximately one section per centimeter of aorta excised) aids in the identification of acute and healed dissections, aortitis, and the extent of cystic meial degeneration.

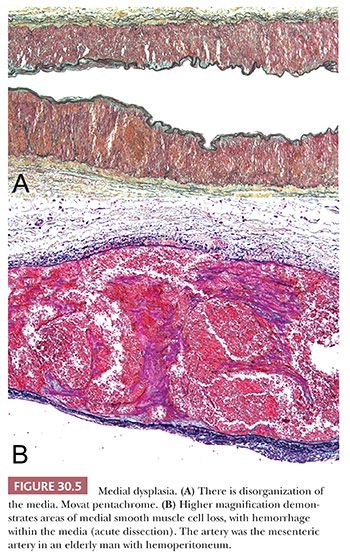
FIBROMUSCULAR DYSPLASIA
Fibromuscular dysplasia is a generic term for a group of structural abnormalities of one or more layers of medium–sized and large arteries that result in aneurysms and dissections of the media (Fig. 30.5). Imaging studies suggest that the disease may be familial in a significant proportion of patients, with an autosomal dominant form of inheritance (8,9). It occurs most often in young to middle-aged white women. Clinical manifestations reflect the arterial bed involved, most commonly hypertension (renal) and stroke (carotid). Dysplasia of the renal arteries and carotid and visceral beds may be localized or part of a systemic process; angiography may be indicated to define the extent of disease in a given patient. The histologic features are not well defined but include disorganization of the media with segmental thinning (classic form of medial dysplasia), diffuse intimal thickening, and primarily adventitial scarring. Mild medial disorganization is characteristic of normal renal arteries, and care should be taken not to overdiagnose fibromuscular dysplasia based on histologic assessment alone.
PERIPHERAL ANEURYSMS AND DISSECTIONS
Aneurysms of the peripheral arteries may be the result of vasculitis (especially polyarteritis nodosa) or septic embolism (infectious aneurysms, especially in patients with infectious endocarditis). More commonly, they are noninflammatory and often of uncertain etiology. Noninflammatory aneurysms may be associated with either medial dysplasia or cystic medial degeneration, and hypertension is a frequent clinical finding. Splenic aneurysms, the most common site for visceral aneurysms, are pathologically heterogeneous and may be the result of medial dysplasia or degeneration, vasculitis, adjacent pancreatitis, portal hypertension, or atherosclerosis. The histologic features are typically obscured by secondary changes of calcification and fibrosis, making a specific diagnosis impossible. In addition to medial dysplasia and medial degeneration, potential causes of peripheral aneurysms include atherosclerotic aneurysms (most common in the leg) and cystic adventitial disease of the popliteal artery.
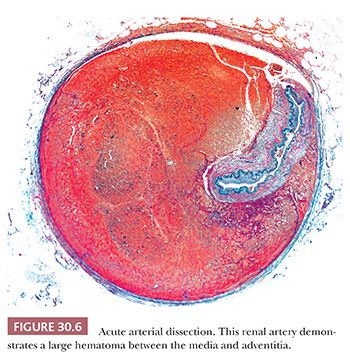
Spontaneous dissections may occur in peripheral arteries with or without preexisting aneurysm (Fig. 30.6). As with aneurysms, predisposing conditions include medial dysplasia and medial degeneration. Spontaneous dissections of the renal arteries often occur in young men without evidence of renal artery dysplasia and may be related to exertion (10).
AMYLOIDOSIS
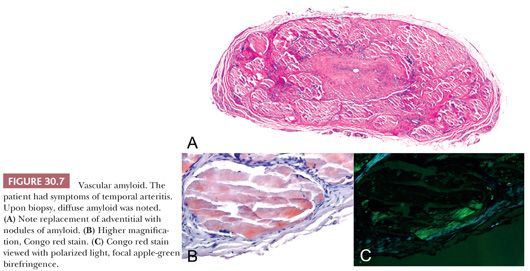
Amyloidosis may result in bleeding, either secondary to coagulopathy or direct involvement of the arterial wall (11). The protein precursor may be free light chains, prealbumin, or AA protein (12). Microvessels may be involved in amyloidosis secondary to β2-microglobulin associated with chronic dialysis. Temporal artery biopsy may occasionally disclose amyloid secondary to amyloid light chain amyloidosis (13), mimicking the clinical symptoms of temporal arteritis (Fig. 30.7).
MEDIAL CALCIFICATION: MÖNCKEBERG AND VASCULAR CALCINOSIS (CALCIPHYLAXIS)
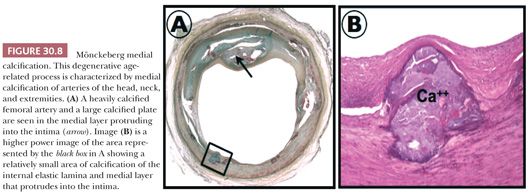
Peripheral vascular disease (PVD) affects over 12 million people in the United States alone. Common pathologic findings in PVD include atherosclerosis and medial calcification. Medial calcification was first described by Mönckeberg in the early twentieth century as a progressive condition found in the peripheral arteries of elderly patients. Calcification classically involves the media and elastic laminae and may progress to circumferential involvement (Fig. 30.8). Medial calcification may also play a role in the progression of PVD and appears to be enhanced by comorbidities such as diabetes mellitus (14).
More severe forms of calcification of the media, which result in a marked giant cell reaction in proximal arteries, extension of calcification into distal vessels, and elevation of acute phase reactants, are termed vascular calcinosis or calciphylaxis. Obstruction of the vessel occurs because of an intimal fibrotic reaction, causing ischemia symptoms or progressive ischemic gangrene. Most patients have renal failure with hyperparathyroidism; calciphylaxis affects 1% to 4% of the population with end-stage renal disease. Radiographs demonstrate diffuse vascular calcifications of vessels of the extremities, but the diagnosis rests on histologic confirmation of calcification in distal arteries and arterioles. Subcutaneous calcium deposits with panniculitis and fat necrosis may sometimes be found. Vascular microthrombi are frequently evident.
ATHEROSCLEROSIS
AORTIC ANEURYSMS
Atherosclerosis of the aorta has been classified into nonprogressive lesions (adaptive intimal thickening [AIT] and fatty streak [FS]). FSs are lipid-rich, grossly yellow, and nonraised. Progressive lesions consist of pathologic intimal thickening (PIT), fibroatheroma, thin-cap fibroatheroma, plaque rupture, and ulcerated plaques. Histologically, PIT consists of smooth muscle cell–rich lesions with lipid pools. Fibroatheromas, by contrast, are raised, firm white lesions with necrotic cores and overlying fibrous caps. Advanced lesions are those with rupture of the fibrous cap and luminal thrombus or ulcerated plaques. Complications of advanced lesions causing clinical symptoms include aneurysm formation, luminal thrombotic obstruction, and atheroembolism. Aortic atherosclerosis is most advanced at the takeoff of arch vessels and the abdominal segment below the renal arteries and above the iliac bifurcation and is often accompanied by iliac disease. The thoracic aorta, especially the descending portion, may also be involved in aneurysmal disease but is less frequent.
The incidence of aortic aneurysms has been increasing (15). The mechanism of why only a fraction of patients with severe aortic atherosclerosis develop aneurysms is unknown, leading to the possibility that other cause may also be involved. White race, hypertension, and smoking are risk factors, and there are likely genetic factors that contribute to increased elastolysis and collagenolysis within the aortic media. High circulating levels of matrix metalloproteinases have also been implicated.
The surgical pathologist will encounter aortic atherosclerosis most commonly as aortic aneurysm repair. The repair of abdominal aortic aneurysms is performed largely to prevent the complication of rupture, which can lead to hemoperitoneum and sudden death. For surgical repair, the wall of the aneurysm is generally left in place, with Dacron conduit interposed between nonaneurysmal areas proximally and distally. The pathologist therefore receives only portions of luminal thrombus, atherosclerotic intima, and inflamed media and adventitia. Today, a larger number of patients have percutaneous aortic stent graft placement as this procedure is less invasive.
CAROTID ENDARTERECTOMY
Carotid endarterectomy is frequently performed in symptomatic patients as well as in asymptomatic patients with severe carotid stenosis. The surgeon attempts to remove the specimen in toto or in multiple pieces. The pathologist should radiograph the specimen, note presence or absence of ruptures or thrombi, and decalcify in ethylenediaminetetraacetic acid if necessary prior to sectioning the plaque transversely for histologic analysis. As with coronary atherectomy, a proportion of patients may have a history of previous endarterectomy, and the indication for the procedure will be recurrence of the disease or restenosis.
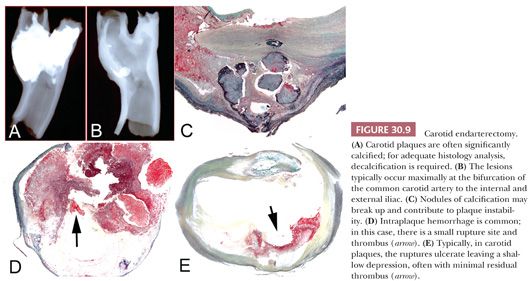
Plaque rupture is the predominant etiology of carotid artery thrombosis in symptomatic patients. Rates of plaque rupture in the carotid (occurring in approximately 70% of patients with stroke) are similar to those reported for the coronary arteries in patients with acute coronary syndromes. In contrast, plaque erosion as a cause of luminal thrombus is quite rare in the carotid but accounts for approximately 35% of thrombotic sudden coronary death (16,17). Of further importance, the carotid distribution is anatomically different from the coronary bed because vessel size is larger and blood flow significantly higher, which may explain the higher rates of distal embolization responsible for strokes or transient ischemic attacks. Therefore, presence of thrombus at the rupture or site of erosion in the carotid artery is only rarely observed and when present is usually small (Fig. 30.9). Although infrequent in the carotid location, disrupted calcified nodule is responsible for approximately 6% to 7% of thrombi. The plaque composition of carotid endarterectomies has been correlated with risk factors. Hypertension and dyslipidemia were the modifiable risk factors mainly correlated with presence of carotid plaque vulnerability and thrombosis (16).
AORTIC OCCLUSIVE DISEASE
Some patients with severe aortic atherosclerosis develop primarily occlusive disease (18), with little if any dilatation distal to the renal arteries. These patients are treated with aortobifemoral bypass grafting and often have embolic symptoms and symptoms of severe PVD. There may be a component of coagulopathy as an etiology for aortoiliac occlusive disease, especially in younger women, and there is an association with atherosclerosis of other arterial beds as well as chronic renal failure.
INFLAMMATORY AORTIC ANEURYSMS
Approximately 10% of patients with abdominal aortic aneurysm demonstrate an inflammatory variant, characterized both grossly as well as histologically (19,20). Male sex and smoking are even stronger risk factors than atherosclerotic aortic aneurysms. Grossly, there is marked thickening of the aneurysm wall, fibrosis of the adjacent retroperitoneum, and adherence of the adjacent structures to the anterior aneurysm wall. Histologically, there is expansion of the adventitia with fibrosis, lymphocytes, and plasma cells (Fig. 30.10).
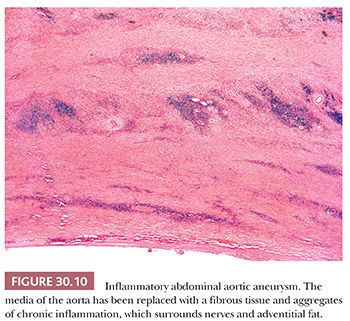
Inflammatory aortic aneurysm is sometimes considered a manifestation of retroperitoneal fibrosis, and purported etiologies include autoimmune disease, possibly triggered by Chlamydia pneumoniae, and immunoglobulin (Ig) G4–related sclerosing disease akin to autoimmune sclerosing pancreatitis. Although there is typically superimposed atherosclerosis, inflammatory aortic aneurysm is considered separately from atherosclerotic disease and is treated more as an isolated vasculitis, with a combination of surgery and anti-inflammatory medications. Inflammation of other vessels is, however, not a feature of inflammatory aortic aneurysm.
It is worth noting that there have been recent significant advances in our understanding of IgG4-related systemic disease (RSD) with the recognition that it may represent an underreported cause of thoracic aortitis, inflammatory abdominal aneurysm, aortic dissection, and retroperitoneal fibrosis (21,22). Histologically, aortic involvement is characterized by an aortitis or periaortitis consisting of a prominent lymphoplasmacytic infiltrate with variable storiform fibrosis. A high percentage (>50%) of the plasma cells stain for IgG4. Clinical evidence of systemic involvement and elevated serum IgG4 levels should be documented in suspected cases. IgG-4 RSD often responds well to a prolonged course of glucocorticoid treatment, underscoring the importance of recognition of this newly described disorder.
VASCULITIS
INTRODUCTION
The term vasculitis means different things in different settings. To the clinician, a diagnosis of vasculitis connotes a systemic autoimmune disease, with a variably chronic course, that has a prominent vascular component. To the pathologist, vasculitis generally denotes the presence of primary vascular inflammation, in the absence of infection, infarction, or other secondary cause. Because most of the systemic vasculitis syndromes have isolated variants that are identical to their systemic counterparts, the pathologic definition of vasculitis is in some sense broader than that of the clinician. Pathologically, it has been stated that certain histologic features (e.g., fibrinoid necrosis) are a prerequisite for the diagnosis of vasculitis. However, it is well known that the histologic findings in patients with known vasculitis syndromes vary by vessel size and type of vasculitis; for example, necrosis is not a typical feature of giant cell vasculitis, and vascular inflammation in nerve or muscle biopsies may not show necrosis in patients with polyarteritis. Therefore, the use of the term vasculitis by the pathologist is generally best qualified by context and explained in a report on what the clinical ramifications may be.
The classification of vasculitis depends on at least three major features: size and type of vessel involved, presence of immune complexes and association with known autoimmune disease, and systemic or localized distribution (23,24). Two major vasculitic syndromes affect elastic arteries: Takayasu disease and giant cell arteritis (Table 30.2). In addition, there is increasing recognition of a third diagnostic category, referred to as nonspecific, idiopathic, or isolated aortitis. “Isolated” aortitis refers to aortitis with histologic features indistinguishable from Takayasu disease and giant cell aortitis but without evidence of systemic disease (25–27). The two features most helpful for distinguishing isolated aortitis from the other forms of ascending aortitis are both clinical rather than microscopic in nature. They include the absence of signs or symptoms of extra-aortic arteritis and the absence of systemic disease. Therefore, complete evaluation with imaging to determine extent of disease and inflammatory markers to determine activity of disease have been advised in patients with isolated aortitis.

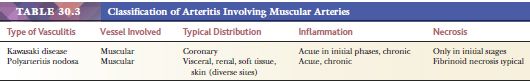
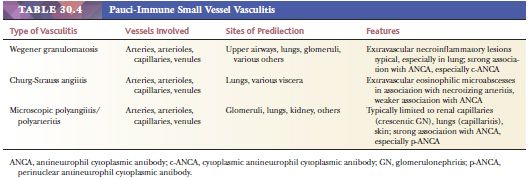
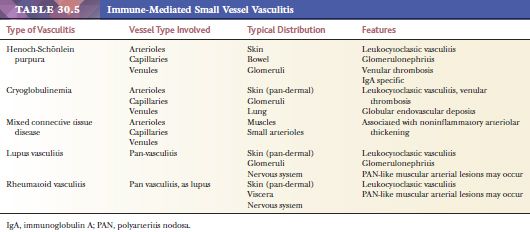
Two vasculitic syndromes affect muscular arteries in the absence of small vessel involvement: polyarteritis nodosa and Kawasaki disease (Table 30.3). A host of vasculitis syndromes affect small vessels (arterioles, capillaries, and venules). These are generally divided into those that are pauci-immune (often antineutrophil cytoplasmic antibody [ANCA] related) (Table 30.4) and those that are caused by immune complex deposition (Table 30.5). The vasculitic syndromes that involve small vessels often affect muscular arteries as well; therefore, histologic features are indistinguishable from polyarteritis if only muscular arteries are sampled histologically.
The pathologist’s role in the diagnosis of vasculitis is often limited, as clinical and serologic evaluation by a rheumatologist is often necessary for a specific diagnosis. Therefore, if the pathologist initially diagnoses vasculitis, it is recommended that the surgical pathology report indicate that further workup be instituted to determine if (a) the process is systemic or isolated and (b) if there are associated autoimmune syndromes. Of course, if the biopsy is performed specifically to rule in or out vasculitis, specifically in the case of temporal artery, nerve, or muscle biopsy, then the pathologist’s role is more straightforward.
TAKAYASU DISEASE
Historically, Takayasu disease was defined as inflammatory peripheral arterial stenoses, first described as ophthalmic artery occlusion. Subsequently, it was determined in autopsy studies from Japan that the inflammatory process involved the aortic arch as well, and gradually, the concept of Takayasu aortitis with aneurysm and stenosis of arch vessels evolved. Eventually, it has become somewhat accepted to use the term for any noninfectious aortitis, in the absence of other autoimmune disease, in virtually any age group (25). The most common initial finding is aortic root disease with aneurysm in the absence of systemic illness. However, clinical criteria for diagnosis include young age range and specific clinical findings indicating autoimmune syndrome and peripheral stenoses. The use of the term Takayasu disease in the medical literature without uniform criteria has resulted in significant confusion in classification of vasculitis of large vessels.
As a unifying concept, Takayasu disease is a necrotizing vasculitis that involves the aorta and branch vessels. In a subset of patients, who are generally women younger than age 50 years, there is diffuse involvement, with extension into peripheral arteries, resulting in systemic autoimmune syndrome with peripheral stenoses. Although not accepted, this subset of patients could be termed Takayasu syndrome. In contrast, patients with Takayasu disease range in age from 20 to more than 80 years, have no gender predilection, and suffer from aortic root dilatation and valve insufficiency without systemic vasculitis (28,29).
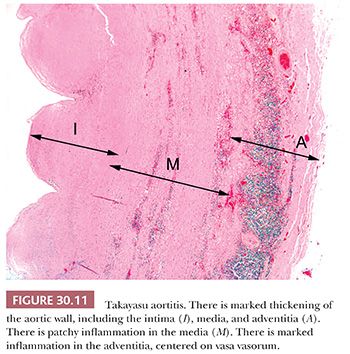
The gross features of aortitis are wrinkling of the intima (so-called tree barking) with variable thickening of the aortic wall (28,29). The histologic features of necrotizing aortitis include zonal medial necrosis rimmed by macrophage giant cells (Fig. 30.11) (26). With healing, there is intimal and adventitial fibrosis. Curiously, healed lesions of the media are relatively devoid of scarring (unlike the fibrosis one sees in the intima and adventitia). In old lesions, there is an accumulation of proteoglycans, which impart an appearance of dysplasia. Extensive sampling with elastic stains to demonstrate the zonal necrosis may be necessary to establish a diagnosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


