24 8 Examination of the spleen Move hand up from right iliac fossa, towards left upper quadrant on expiration. Place your left hand around patient’s lower ribs and approach costal margin to pull spleen forwards. To help palpate small spleens, roll the patient on to the right side and examine as before. Disorders of the blood cover a wide spectrum of illnesses, ranging from some of the most common disorders affecting mankind (anaemias) to relatively rare conditions such as leukaemias and congenital coagulation disorders. Although the latter are uncommon, advances in cellular and molecular biology have had major impacts on their diagnosis, treatment and prognosis. Haematological changes occur as a consequence of diseases affecting any system and give important information in the diagnosis and monitoring of many conditions. Blood flows throughout the body in the vascular system, and consists of: • red cells, which transport oxygen from the lungs to the tissues • white cells, which defend against infection • platelets, which interact with blood vessels and clotting factors to maintain vascular integrity and prevent bleeding • plasma, which contains proteins with many functions, including antibodies and coagulation factors. Bone marrow contains a range of immature haematopoietic precursor cells and a storage pool of mature cells for release at times of increased demand. Haematopoietic cells interact closely with surrounding connective tissue stroma, made up of reticular cells, macrophages, fat cells, blood vessels and nerve fibres (Fig. 24.1). In normal marrow, nests of red cell precursors cluster around a central macrophage, which provides iron and also phagocytoses nuclei from red cells prior to their release into the circulation. Megakaryocytes are large cells which produce and release platelets into vascular sinuses. White cell precursors are clustered next to the bone trabeculae; maturing cells migrate into the marrow spaces towards the vascular sinuses. Plasma cells are antibody-secreting mature B cells which normally represent less than 5% of the marrow population and are scattered throughout the intertrabecular spaces. All blood cells are derived from pluripotent haematopoietic stem cells. These comprise only 0.01% of the total marrow cells, but they can self-renew (i.e. make more stem cells) or differentiate to produce a hierarchy of lineage-committed stem cells. The resulting primitive progenitor cells cannot be identified morphologically, so they are named according to the types of cell (or colony) they form during cell culture experiments. CFU–GM (colony-forming unit – granulocyte, monocyte) are stem cells that produce granulocytic and monocytic lines, CFU–E produce erythroid cells, and CFU–Meg produce megakaryocytes and ultimately platelets (Fig. 24.2). The bone marrow also contains stem cells which can differentiate into non-haematological cells, such as nerve, skeletal muscle, cardiac muscle, liver and blood vessel endothelium. This is termed stem-cell plasticity and may have exciting clinical applications in the future (Ch. 3). Red cell precursors formed in the bone marrow from the erythroid (CFU–E) progenitor cells are called erythroblasts or normoblasts (Fig. 24.3). These divide and acquire haemoglobin, which turns the cytoplasm pink; the nucleus condenses and is extruded from the cell. The first non-nucleated red cell is a reticulocyte, which still contains ribosomal material in the cytoplasm, giving these large cells a faint blue tinge (‘polychromasia’). Reticulocytes lose their ribosomal material and mature over 3 days, during which time they are released into the circulation. Increased numbers of circulating reticulocytes (reticulocytosis) reflect increased erythropoiesis. Proliferation and differentiation of red cell precursors is stimulated by erythropoietin, a polypeptide hormone produced by renal interstitial peritubular cells in response to hypoxia. Failure of erythropoietin production in patients with renal failure (p. 478) causes anaemia, which can be treated with exogenous recombinant erythropoietin. Normal mature red cells circulate for about 120 days. They are 8 µm biconcave discs lacking a nucleus but filled with haemoglobin, which delivers oxygen to the tissues. In order to pass through the smallest capillaries, the red cell membrane is deformable, with a lipid bilayer to which a ‘skeleton’ of filamentous proteins is attached via special linkage proteins (Fig. 24.4). Inherited abnormalities of any of these proteins result in loss of membrane as cells pass through the spleen, and the formation of abnormally shaped red cells called spherocytes or elliptocytes (see Fig. 24.8D, p. 999). Red cells are exposed to osmotic stress in the pulmonary and renal circulation; in order to maintain homeostasis, the membrane contains ion pumps, which control intracellular levels of sodium, potassium, chloride and bicarbonate. In the absence of mitochondria, the energy for these functions is provided by anaerobic glycolysis and the pentose phosphate pathway in the cytosol. Membrane glycoproteins inserted into the lipid bilayer also form the antigens recognised by blood grouping (see Fig. 24.4). The ABO and Rhesus systems are the most commonly recognised (p. 1012), but over 400 blood group antigens have been described. Haemoglobin is a protein specially adapted for oxygen transport. It is composed of four globin chains, each surrounding an iron-containing porphyrin pigment molecule termed haem. Globin chains are a combination of two alpha and two non-alpha chains; haemoglobin A (αα/ββ) represents over 90% of adult haemoglobin, whereas haemoglobin F (αα/γγ) is the predominant type in the fetus. Each haem molecule contains a ferrous ion (Fe2+), to which oxygen reversibly binds; the affinity for oxygen increases as successive oxygen molecules bind. When oxygen is bound, the beta chains ‘swing’ closer together; they move apart as oxygen is lost. In the ‘open’ deoxygenated state, 2,3 diphosphoglycerate (DPG), a product of red cell metabolism, binds to the haemoglobin molecule and lowers its oxygen affinity. These complex interactions produce the sigmoid shape of the oxygen dissociation curve (Fig. 24.5). The position of this curve depends upon the concentrations of 2,3 DPG, H+ ions and CO2; increased levels shift the curve to the right and cause oxygen to be released more readily, e.g. when red cells reach hypoxic tissues. Haemoglobin F is unable to bind 2,3 DPG and has a left-shifted oxygen dissociation curve, which, together with the low pH of fetal blood, ensures fetal oxygenation. Genetic mutations affecting the haem-binding pockets of globin chains or the ‘hinge’ interactions between globin chains result in haemoglobinopathies or unstable haemoglobins. Alpha globin chains are produced by two genes on chromosome 16, and beta globin chains by a single gene on chromosome 11; imbalance in the production of globin chains results in the thalassaemias (p. 1034). Defects in haem synthesis cause the porphyrias (p. 458). White cells or leucocytes in the blood consist of granulocytes (neutrophils, eosinophils and basophils), monocytes and lymphocytes (see Fig. 24.12, p. 1004). Granulocytes and monocytes are formed from bone marrow CFU–GM progenitor cells during myelopoiesis. The first recognisable granulocyte in the marrow is the myeloblast, a large cell with a small amount of basophilic cytoplasm and a primitive nucleus with open chromatin and nucleoli. As the cells divide and mature, the nucleus segments and the cytoplasm acquires specific neutrophilic, eosinophilic or basophilic granules (see Fig. 24.3). This takes about 14 days. The cytokines G–CSF, GM–CSF and M–CSF are involved in the production of myeloid cells, and G–CSF can be used clinically to hasten recovery of blood neutrophil counts after chemotherapy. Neutrophils, the most common white blood cells in the blood of adults, are 10–14 µm in diameter, with a multilobular nucleus containing 2–5 segments and granules in their cytoplasm. Their main function is to recognise, ingest and destroy foreign particles and microorganisms (p. 72). A large storage pool of mature neutrophils exists in the bone marrow. Every day, some 1011 neutrophils enter the circulation, where cells may be circulating freely or attached to endothelium in the marginating pool. These two pools are equal in size; factors such as exercise or catecholamines increase the number of cells flowing in the blood. Neutrophils spend 6–10 hours in the circulation before being removed, principally by the spleen. Alternatively, they pass into the tissues and either are consumed in the inflammatory process or undergo apoptotic cell death and phagocytosis by macrophages. Eosinophils represent 1–6% of the circulating white cells. They are a similar size to neutrophils but have a bilobed nucleus and prominent orange granules on Romanowsky staining. Eosinophils are phagocytic and their granules contain a peroxidase capable of generating reactive oxygen species and proteins involved in the intracellular killing of protozoa and helminths (p. 311). They are also involved in allergic reactions (e.g. atopic asthma, p. 666; see also p. 89). Monocytes are the largest of the white cells, with a diameter of 12–20 µm and an irregular nucleus in abundant pale blue cytoplasm containing occasional cytoplasmic vacuoles. These cells circulate for a few hours and then migrate into tissue, where they become macrophages, Kupffer cells or antigen-presenting dendritic cells. The former phagocytose debris, apoptotic cells and microorganisms (see Box 4.1, p. 74). Lymphocytes are derived from pluripotent haematopoietic stem cells in the bone marrow. There are two main types: T cells (which mediate cellular immunity) and B cells (which mediate humoral immunity) (p. 77). Lymphoid cells that migrate to the thymus develop into T cells, whereas B cells develop in the bone marrow. Blood must be maintained in a fluid state in order to function as a transport system, but must be able to solidify to form a clot following vascular injury in order to prevent excessive bleeding, a process known as haemostasis. Successful haemostasis is localised to the area of tissue damage and is followed by removal of the clot and tissue repair. This is achieved by complex interactions between the vascular endothelium, platelets, coagulation factors, natural anticoagulants and fibrinolytic enzymes (Fig. 24.6). Dysfunction of any of these components may result in haemorrhage or thrombosis. Under normal conditions platelets are discoid, with a diameter of 2–4 µm (Fig. 24.7). The surface membrane invaginates to form a tubular network, the canalicular system, which provides a conduit for the discharge of the granule content following platelet activation. Drugs which inhibit platelet function and thrombosis include aspirin (cyclo-oxygenase inhibitor), clopidogrel (adenosine diphosphate (ADP)-mediated activation inhibitor), dipyridamole (phosphodiesterase inhibitor), and the IIb/IIIa inhibitors abciximab, tirofiban and eptifibatide (which prevent fibrinogen binding; p. 594). The coagulation system consists of a cascade of soluble inactive zymogen proteins designated by Roman numerals. When proteolytically cleaved and activated, each is capable of activating one or more components of the cascade. Activated factors are designated by the suffix ‘a’. Some of these reactions require phospholipid and calcium. Coagulation occurs by two pathways: it is initiated by the extrinsic (or tissue factor) pathway and amplified by the ‘intrinsic pathway’ (see Fig. 24.6). Clotting factors are synthesised by the liver, although factor V is also produced by platelets and endothelial cells. Factors II, VII, IX and X require post-translational carboxylation to allow them to participate in coagulation. The carboxylase enzyme responsible for this in the liver is vitamin K-dependent. Vitamin K is converted to an epoxide in this reaction and must be reduced to its active form by a reductase enzyme. This reductase is inhibited by warfarin, and this is the basis of the anticoagulant effect of coumarins (p. 1019). Congenital (e.g. haemophilia) and acquired (e.g. liver failure) causes of coagulation factor deficiency are associated with bleeding. To obtain a full blood count (FBC), anticoagulated blood is processed through automated blood analysers which use a variety of technologies (particle-sizing, radiofrequency and laser instrumentation) to measure the haematological parameters. These include numbers of circulating cells, the proportion of whole blood volume occupied by red cells (the haematocrit, Hct), and the red cell indices which give information about the size of red cells (mean cell volume, MCV) and the amount of haemoglobin present in the red cells (mean cell haemoglobin, MCH). Blood analysers can differentiate types of white blood cell and give automated counts of neutrophils, lymphocytes, monocytes, eosinophils and basophils. It is important to appreciate, however, that a number of conditions can lead to spurious results (Box 24.1). The reference ranges for a number of common haematological parameters in adults are given in Chapter 29. Although technical advances in full blood count analysers have resulted in fewer blood samples requiring manual examination, scrutiny of blood components prepared on a microscope slide (the ‘blood film’) can often yield valuable information (Box 24.2 and Fig. 24.8). Analysers cannot identify abnormalities of red cell shape and content (e.g. Howell–Jolly bodies, basophilic stippling, malaria parasites) or fully define abnormal white cells such as blasts. In adults, bone marrow for examination is usually obtained from the posterior iliac crest. After a local anaesthetic, marrow can be sucked out from the medullary space, stained and examined under the microscope (bone marrow aspirate). In addition, a core of bone may be removed (trephine biopsy), fixed and decalcified before sections are cut for staining (Fig. 24.9). A bone marrow aspirate is used to assess the composition and morphology of haematopoietic cells or abnormal infiltrates. Further investigations may be performed, such as cell surface marker analysis (immunophenotyping), chromosome and molecular studies to assess malignant disease, or marrow culture for suspected tuberculosis. A trephine biopsy is superior for assessing marrow cellularity, marrow fibrosis, and infiltration by abnormal cells such as metastatic carcinoma. In patients with clinical evidence of a bleeding disorder (p. 991), there are recommended screening tests (Box 24.3). (DIC = disseminated intravascular coagulation) Coagulation tests measure the time to clot formation in vitro in a plasma sample after the clotting process is initiated by activators and calcium. The result of the test sample is compared with normal controls. The tissue factor (‘extrinsic’) pathway (see Fig 24.6) is assessed by the prothrombin time (PT), and the ‘intrinsic’ pathway by the activated partial thromboplastin time (APTT), sometimes known as the partial thromboplastin time with kaolin (PTTK). Coagulation is delayed by deficiencies of coagulation factors and by the presence of inhibitors of coagulation, such as heparin. The approximate reference ranges and causes of abnormalities are shown in Box 24.3. If both the PT and APTT are prolonged, this indicates either deficiency or inhibition of the final common pathway (which includes factors X, V, prothrombin and fibrinogen) or global coagulation factor deficiency involving more than one factor, as occurs in disseminated intravascular coagulation (DIC, pp. 201 and 1055). Further specific tests may be performed based on interpretation of the clinical scenario and results of these screening tests. A mixing test with normal plasma allows differentiation between a coagulation factor deficiency (the prolonged time corrects) and the presence of an inhibitor of coagulation (the prolonged time does not correct); the latter may be chemical (heparins) or an antibody (most often a lupus anticoagulant but occasionally a specific inhibitor of one of the coagulation factors, typically factor VIII). Von Willebrand disease may present with a normal APTT; further investigation of suspected cases is detailed on page 1053. Coagulation screening tests are also performed in patients with suspected DIC, when clotting factors and platelets are consumed, resulting in thrombocytopenia and prolonged PT and APTT. In addition, there is evidence of active coagulation with consumption of fibrinogen and generation of fibrin degradation products (D-dimers). Note, however, that fibrinogen is an acute phase protein which may also be elevated in inflammatory disease (p. 82). Measurement of plasma levels of D-dimers derived from fibrin degradation is useful in excluding the diagnosis of active venous thrombosis in some patients (see Fig. 24.15, p. 1010). A variety of tests exist which may help to explain an underlying propensity to thrombosis, especially venous thromboembolism (thrombophilia) (Box 24.4). Examples of possible indications for testing are given in Box 24.5. In most patients, the results do not affect clinical management (p. 1054) but they may influence the duration of anticoagulation (e.g. antiphospholipid antibodies, p. 1055), justify family screening in inherited thrombophilias (p. 1054), or suggest additional management strategies to reduce thrombosis risk (e.g. in myeloproliferative disease and paroxysmal nocturnal haemoglobinuria; p. 1031). Anticoagulants can interfere with some of these assays; for example, warfarin reduces protein C and S levels and affects measurement of lupus anticoagulant, while heparin interferes with antithrombin and lupus anticoagulant assays. Therefore these tests, when required, should be performed when the patient is not taking anticoagulants. *Antiphospholipid antibodies should be sought where clinical criteria for antiphospholipid syndrome (APS) are fulfilled (p. 1055). Thrombophilia testing may explain the diagnosis without necessarily affecting management. • Blood cell counts and film components: not altered in general by ageing alone, although haemoglobin concentrations fall with increasing age. • Ratio of bone marrow cells to marrow fat: falls. • Neutrophils: maintained throughout life, although leucocytes may be less readily mobilised by bacterial invasion in old age. • Lymphocytes: functionally compromised by age due to a T cell-related defect in cell-mediated immunity. • Clotting factors: no major changes, although mild congenital deficiencies may be first noticed in old age. • Erythrocyte sedimentation rate (ESR): raised above the reference range, but usually in association with chronic or subacute disease. In truly healthy older people, the ESR range is very similar to that in younger people. Anaemia refers to a state in which the level of haemoglobin in the blood is below the reference range appropriate for age and sex. Other factors, including pregnancy and altitude, also affect haemoglobin levels and must be taken into account when considering whether an individual is anaemic. The clinical features of anaemia reflect diminished oxygen supply to the tissues (p. 991). A rapid onset of anaemia (e.g. due to blood loss) causes more profound symptoms than a gradually developing anaemia. Individuals with cardiorespiratory disease are more susceptible to symptoms of anaemia. The clinical assessment and investigation of anaemia should gauge its severity and define the underlying cause (Box 24.7).
Blood disease
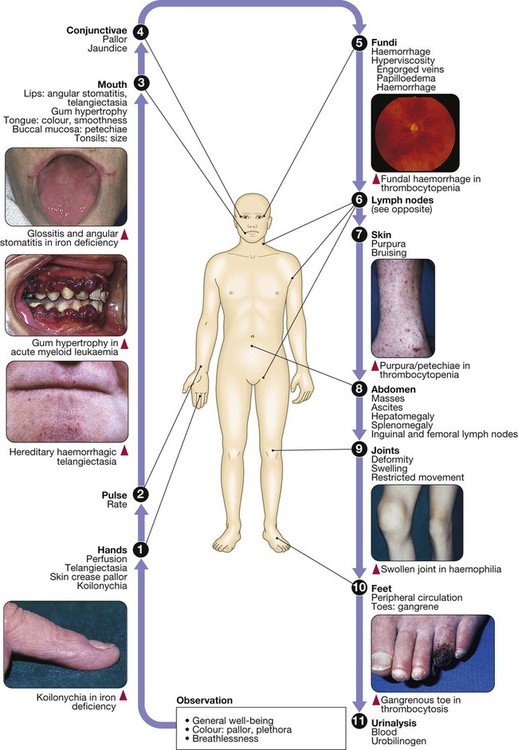
Clinical examination in blood disease
Bleeding
 Bleeding
Bleeding
Functional anatomy and physiology
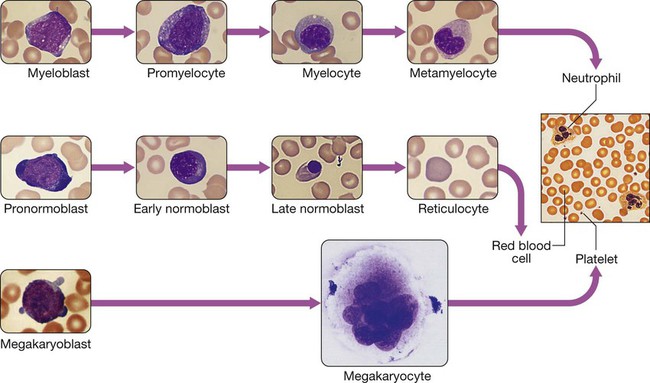
Haematopoiesis
Stem cells
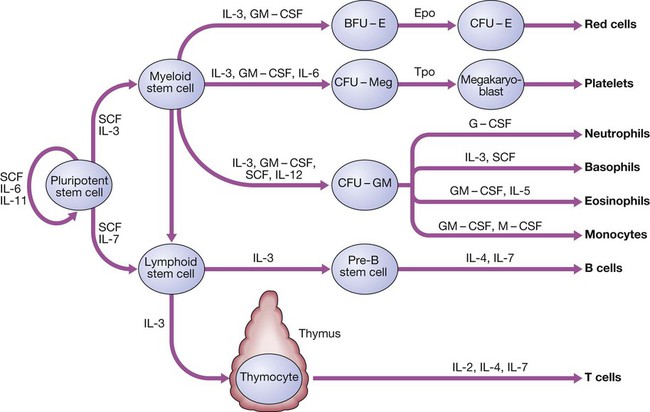
(BFU-E = burst-forming unit – erythroid; CFU–E = colony-forming unit – erythroid; CFU–GM = colony-forming unit – granulocyte, monocyte; CFU–Meg = colony-forming unit – megakaryocyte; Epo = erythropoietin; G–CSF = granulocyte–colony-stimulating factor; GM–CSF = granulocyte, macrophage–colony-stimulating factor; IL = interleukin; M–CSF = macrophage–colony-stimulating factor; SCF = stem cell factor; Tpo = thrombopoietin)
Blood cells and their functions
Red cells
The image on the right is normal blood film.
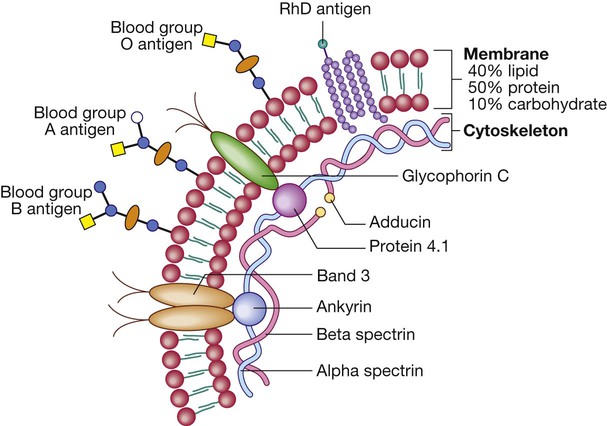
Red cell membrane flexibility is conferred by attachment of cytoskeletal proteins. Important transmembrane proteins include band 3 (an ion transport channel) and glycophorin (involved in cytoskeletal attachment and gas exchange, and a receptor for Plasmodium falciparum in malaria). Antigens on the red blood cell determine an individual’s blood group. There are about 22 blood group systems (groups of carbohydrate or protein antigens controlled by a single gene or by multiple closely linked loci); the most important clinically are the ABO and Rhesus (Rh) systems (p. 1012). The ABO genetic locus has three main allelic forms: A, B and O. The A and B alleles encode glycosyltransferases that introduce N-acetylgalactosamine (open circle) and D-galactose (blue circle), respectively, on to antigenic carbohydrate molecules on the membrane surface. People with the O allele produce an O antigen, which lacks either of these added sugar groups. Rh antigens are transmembrane proteins.
Haemoglobin
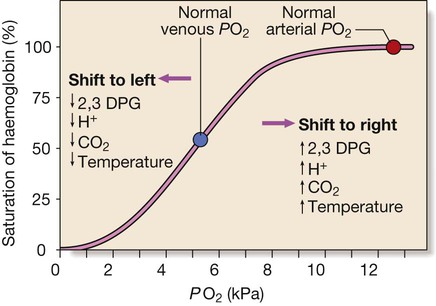
Factors are listed which shift the curve to the right (more oxygen released from blood) and to the left (less oxygen released) at given PO2. (To convert kPa to mmHg, multiply by 7.5.)
White cells
Neutrophils
Eosinophils
Monocytes
Lymphocytes
Haemostasis
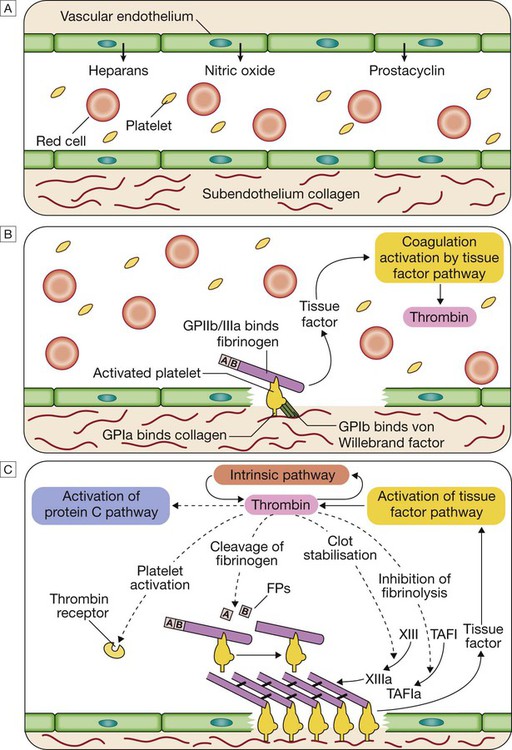
A Stage 1 Pre-injury conditions encourage flow The vascular endothelium produces substances (including nitric oxide, prostacyclin and heparans) to prevent adhesion of platelets and white cells to the vessel wall. Platelets and coagulation factors circulate in a non-activated state.
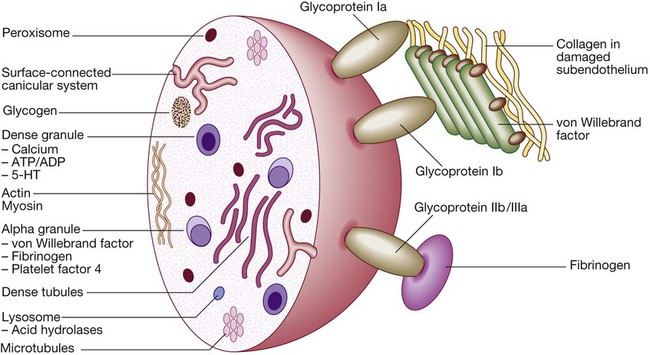
B Stage 2 Early haemostatic response: platelets adhere; coagulation is activated. At the site of injury, the endothelium is breached, exposing subendothelial collagen. Small amounts of tissue factor (TF) are released. Platelets bind to collagen via a specific receptor, glycoprotein Ia (GPIa), causing a change in platelet shape and its adhesion to the area of damage by the binding of other receptors (GPIb and GPIIb/IIIa) to von Willebrand factor and fibrinogen, respectively. Coagulation is activated by the tissue factor (extrinsic) pathway, generating small amounts of thrombin.
C Stage 3 Fibrin clot formation: platelets become activated and aggregate; fibrin formation is supported by the platelet membrane; stable fibrin clot forms. The adherent platelets are activated by many pathways, including binding of adenosine diphosphate (ADP), collagen, thrombin and adrenaline (epinephrine) to surface receptors. The cyclo-oxygenase pathway converts arachidonic acid from the platelet membrane into thromboxane A2, which causes aggregation of platelets. Activation of the platelets results in release of the platelet granule contents, enhancing coagulation further (see Fig. 24.7). Thrombin plays a key role in the control of coagulation: the small amount generated via the TF pathway massively amplifies its own production; the ‘intrinsic’ pathway becomes activated and large amounts of thrombin are generated. Thrombin directly causes clot formation by cleaving fibrinopeptides (FP) from fibrinogen to produce fibrin. Fibrin monomers are cross-linked by factor XIII, which is also activated by thrombin. Having had a key role in clot formation and stabilisation, thrombin then starts to regulate clot formation in two main ways: (a) activation of the protein C (PC) pathway (a natural anticoagulant), which reduces further coagulation; (b) activation of thrombin-activatable fibrinolysis inhibitor (TAFI), which inhibits fibrinolysis (see D and E).
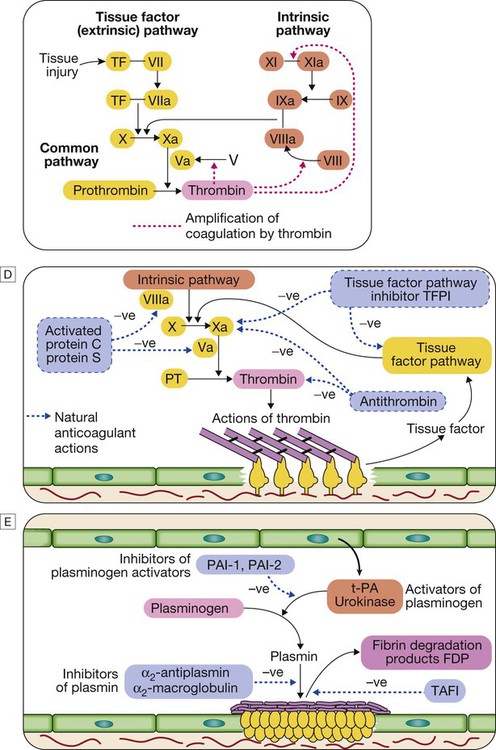
D Stage 4 Limiting clot formation: natural anticoagulants reverse activation of coagulation factors. Once haemostasis has been secured, the propagation of clot is curtailed by anticoagulants. Antithrombin is a serine protease inhibitor synthesised by the liver, which destroys activated factors such as XIa, Xa and thrombin (IIa). Its major activity against thrombin and Xa is enhanced by heparin and fondaparinux, explaining their anticoagulant effect. Tissue factor pathway inhibitor (TFPI) binds to and inactivates VIIa and Xa. Activation of PC occurs following binding of thrombin to membrane-bound thrombomodulin; activated protein C (aPC) binds to its co-factor protein S (PS), and cleaves Va and VIIIa. PC and PS are vitamin K-dependent and are depleted by coumarin anticoagulants such as warfarin.
E Stage 5 Fibrinolysis: plasmin degrades fibrin to allow vessel recanalisation and tissue repair. The insoluble clot needs to be broken down for vessel recanalisation. Plasmin, the main fibrinolytic enzyme, is produced when plasminogen is activated, e.g. by tissue plasminogen activator (t-PA) or urokinase in the clot. Plasmin hydrolyses the fibrin clot, producing fibrin degradation products, including the D-dimer. This process is highly regulated; the plasminogen activators are controlled by an inhibitor called plasminogen activator inhibitor (PAI), the activity of plasmin is inhibited by α2-antiplasmin and α2-macroglobulin, and fibrinolysis is further inhibited by the thrombin-activated TAFI.
Platelets
Clotting factors
Investigation of diseases of the blood
The full blood count
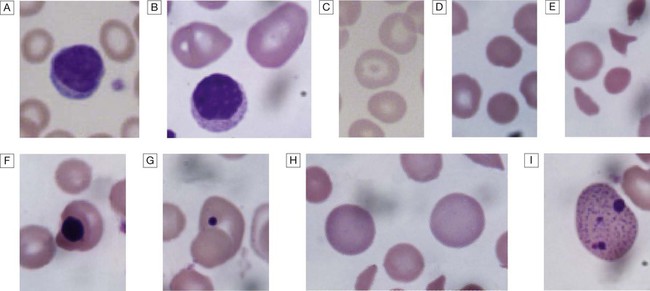
Blood film examination
Bone marrow examination
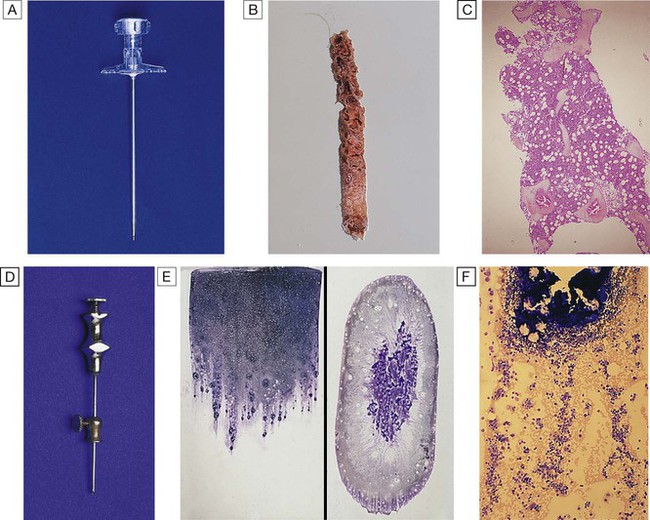
A Trephine biopsy needle. B Macroscopic appearance of a trephine biopsy. C Microscopic appearance of stained section of trephine. D Bone marrow aspirate needle. E Stained macroscopic appearance of marrow aspirate: smear (left) and squash (right). F Microscopic appearance of stained marrow particles and trails of haematopoietic cells.
Investigation of coagulation
Bleeding disorders
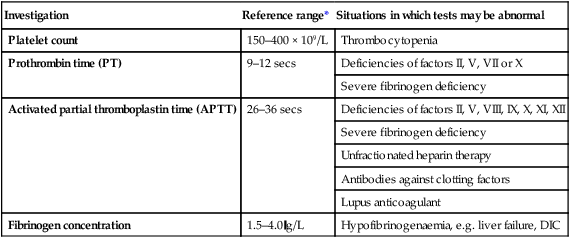
 24.3 Coagulation screening tests
24.3 Coagulation screening tests
Investigation
Reference range*
Situations in which tests may be abnormal
Platelet count
150–400 × 109/L
Thrombocytopenia
Prothrombin time (PT)
9–12 secs
Deficiencies of factors II, V, VII or X
Severe fibrinogen deficiency
Activated partial thromboplastin time (APTT)
26–36 secs
Deficiencies of factors II, V, VIII, IX, X, XI, XII
Severe fibrinogen deficiency
Unfractionated heparin therapy
Antibodies against clotting factors
Lupus anticoagulant
Fibrinogen concentration
1.5–4.0 g/L
Hypofibrinogenaemia, e.g. liver failure, DIC
Thrombotic disorders
 24.5 Indications for thrombophilia testing*
24.5 Indications for thrombophilia testing*

 24.6 Haematological investigations in old age
24.6 Haematological investigations in old age
Presenting problems in blood disease
Anaemia
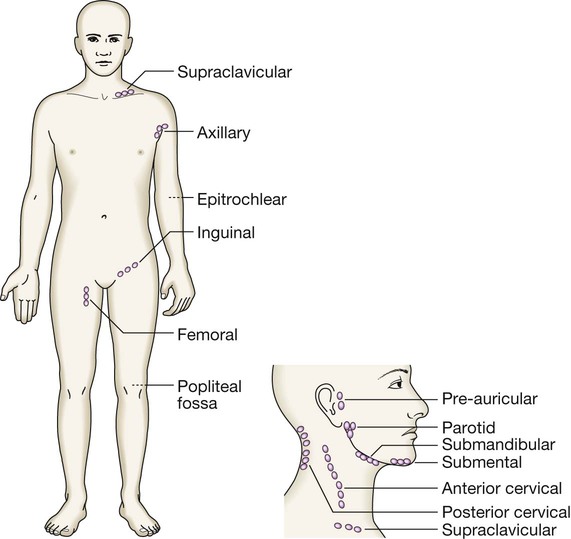
 Lymphadenopathy
Lymphadenopathy Characteristics of the spleen
Characteristics of the spleen Anaemia
Anaemia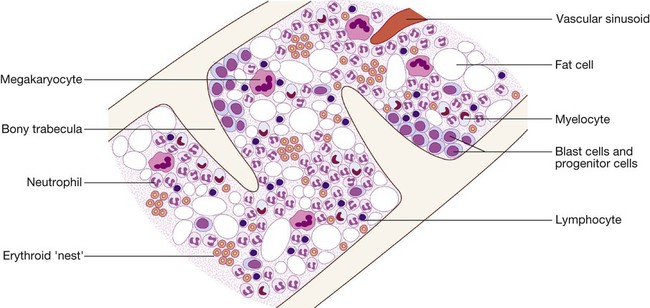
 24.1
24.1 24.2
24.2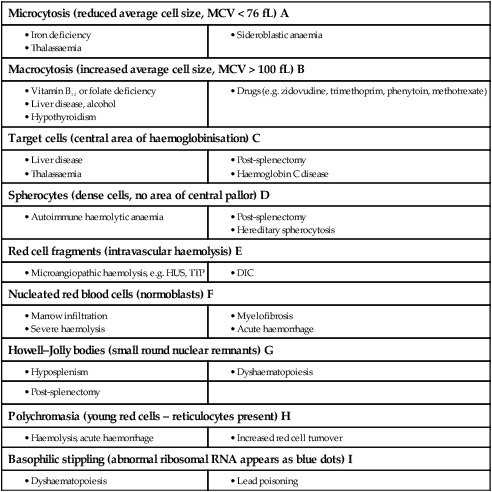
 24.4
24.4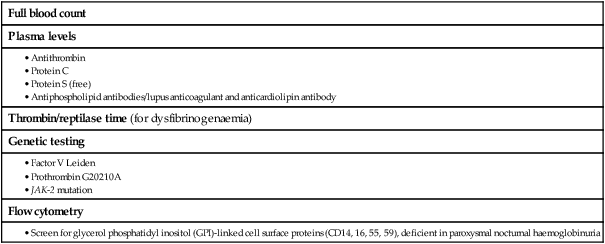
 24.7
24.7


