Blood Coagulation and Anticoagulant, Fibrinolytic, and Antiplatelet Drugs
Blood must remain fluid within the vasculature and yet clot quickly when exposed to subendothelial surfaces at sites of vascular injury. Under normal circumstances, a delicate balance between coagulation and fibrinolysis prevents both thrombosis and hemorrhages. Alteration of this balance in favor of coagulation results in thrombosis. Thrombi, composed of platelet aggregates, fibrin, and trapped red blood cells, can form in arteries or veins. Antithrombotic drugs used to treat thrombosis include antiplatelet drugs, which inhibit platelet activation or aggregation; anticoagulants, which attenuate fibrin formation; and fibrinolytic agents, which degrade fibrin. All antithrombotic drugs increase the risk of bleeding.
This chapter reviews the agents commonly used for controlling blood fluidity, including:
• The parenteral anticoagulant heparin and its derivatives, which activate a natural inhibitor of coagulant proteases
• The coumarin anticoagulants, which block multiple steps in the coagulation cascade
• Fibrinolytic agents, which degrade thrombi
• Antiplatelet agents, including aspirin, thienopyridines, and glycoprotein (GP) IIb/IIIa inhibitors
OVERVIEW OF HEMOSTASIS: PLATELET FUNCTION, BLOOD COAGULATION, AND FIBRINOLYSIS
Hemostasis is the cessation of blood loss from a damaged vessel. Platelets first adhere to macromolecules in the subendothelial regions of the injured blood vessel, where they become activated. Adherent platelets release substances that activate nearby platelets, thereby recruiting them to the site of injury. Activated platelets then aggregate to form the primary hemostatic plug.
Vessel wall injury also exposes tissue factor (TF), which initiates the coagulation system. Platelets enhance activation of the coagulation system by providing a surface onto which clotting factors assemble and by releasing stored clotting factors. This results in a burst of thrombin (factor IIa) generation. Thrombin then converts fibrinogen to fibrin and amplifies platelet activation and aggregation.
Later, as wound healing occurs, the platelet aggregates and fibrin clots are degraded. The processes of platelet aggregation and blood coagulation are summarized in Figures 30–1 and 30–2 (see also the animation on the Goodman & Gilman website). The pathway of clot removal, fibrinolysis, is shown in Figure 30–3, along with sites of action of fibrinolytic agents. Coagulation involves a series of zymogen activation reactions, as shown in Figure 30–2. At each stage, a precursor protein, or zymogen, is converted to an active protease by cleavage of 1 or more peptide bonds in the precursor molecule. The final protease generated is thrombin.
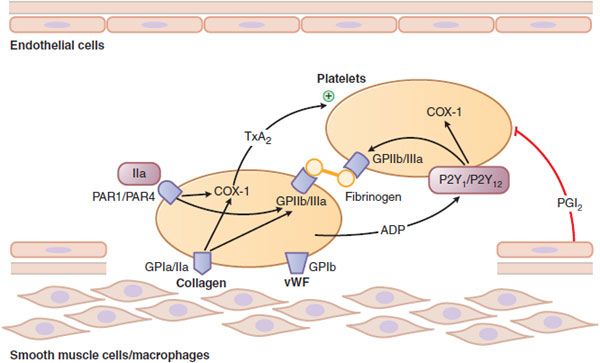
Figure 30–1 Platelet adhesion and aggregation. GPIa/IIa and GPIb are platelet receptors that bind to collagen and von Willebrand factor (vWF), causing platelets to adhere to the subendothelium of a damaged blood vessel. PAR1 and PAR4 are protease-activated receptors that respond to thrombin (IIa); P2Y1 and P2Y12 are receptors for ADP; when stimulated by agonists, these receptors activate the fibrinogen-binding protein GPIIb/IIIa and cyclooxygenase-1 (COX-1) to promote platelet aggregation and secretion. Thromboxane A2 (TxA2) is the major product of COX-1 involved in platelet activation. Prostaglandin I2 (prostacyclin, PGI2), synthesized by endothelial cells, inhibits platelet activation.
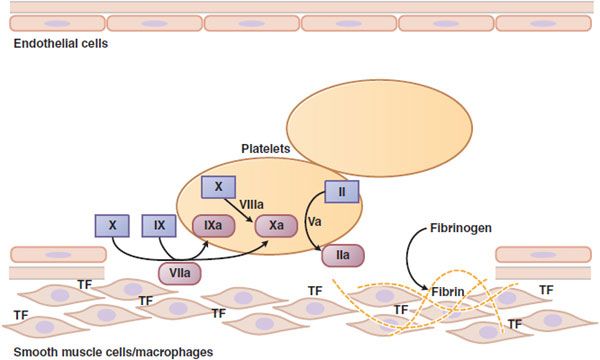
Figure 30–2 Major reactions of blood coagulation. Shown are interactions among proteins of the “extrinsic” (tissue factor and factor VII), “intrinsic” (factors IX and VIII), and “common” (factors X, V, and II) coagulation pathways that are important in vivo. Boxes enclose the coagulation factor zymogens (indicated by Roman numerals); the rounded boxes represent the active proteases. TF, tissue factor. Activated coagulation factors are followed by the letter “a”: II, prothrombin; IIa, thrombin.
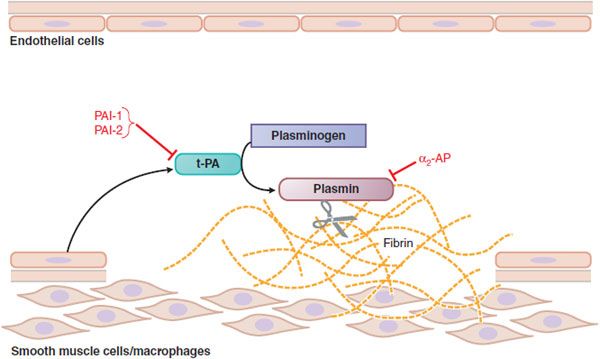
Figure 30–3 Fibrinolysis. Endothelial cells secrete tissue plasminogen activator (t-PA) at sites of injury. t-PA binds to fibrin and converts plasminogen to plasmin, which digests fibrin. Plasminogen activator inhibitors-1 and -2 (PAI-1, PAI-2) inactivate t-PA; α2-antiplasmin (α2-AP) inactivates plasmin.
CONVERSION OF FIBRINOGEN TO FIBRIN. Fibrinogen, a 340,000-Da protein, is a dimer, each half of which consists of 3 pairs of polypeptide chains (designated Aα, Bβ, and γ). Disulfide bonds covalently link the chains and the 2 halves of the molecule together. Thrombin converts fibrinogen to fibrin monomers by releasing fibrinopeptide A (a 16–amino acid fragment) and fibrinopeptide B (a 14–amino acid fragment) from the amino termini of the Aα and Bβ chains, respectively. Removal of the fibrinopeptides creates new amino termini, which fit into preformed holes on other fibrin monomers to form a fibrin gel, which is the end point of in vitro tests of coagulation (see “Coagulation in vitro”). Initially, the fibrin monomers are bound to each other noncovalently. Subsequently, factor XIII, a transglutaminase that is activated by thrombin, catalyzes interchain covalent cross-links between adjacent fibrin monomers, which enhance the strength of the clot.
STRUCTURE OF COAGULATION FACTORS. In addition to factor XIII, the coagulation factors include factors II (prothrombin), VII, IX, X, XI, XII, and prekallikrein. A stretch of ~200 amino acid residues at the carboxyl-termini of each of these zymogens exhibits homology to trypsin and contains the active site of the proteases. In addition, 9-12 glutamate residues near the amino termini of factors II, VII, IX, and X are converted to t-carboxyglutamate (Gla) residues that bind Ca2+ and are necessary for the coagulant activities of these proteins.
NONENZYMATIC PROTEIN COFACTORS. Factors V and VIII serve as cofactors. Factor VIII circulates in plasma bound to von Willebrand factor. Factor V circulates in plasma, is stored in platelets in a partially activated form, and is released when platelets are activated. Thrombin cleaves factors V and VIII to yield activated cofactors (factors Va and VIIIa).
Factors Va and VIIIa serve as cofactors by binding to the surface of activated platelets and acting as receptors for factors Xa and IXa, respectively. The activated cofactors also help localize prothrombin and factor X, the respective substrates for these enzymes, on the activated platelet surface. These coagulation factor complexes increase the catalytic efficiency of factors Xa and IXa by ~109-fold.
TF is a nonenzymatic lipoprotein cofactor; it initiates coagulation by enhancing the catalytic efficiency of factor VIIa. Not normally present on blood-contacting cells, TF is constitutively expressed on the surface of subendothelial smooth muscle cells and fibroblasts, which are exposed when the vessel wall is damaged. Another plasma protein, high-molecular-weight kininogen, also serves as a cofactor.
ACTIVATION OF PROTHROMBIN. By cleaving 2 peptide bonds on prothrombin, factor Xa converts it to thrombin. In the presence of factor Va, a negatively charged phospholipid surface, and Ca2+, factor Xa activates prothrombin with 109-fold greater efficiency. The maximal rate of activation only occurs when prothrombin and factor Xa contain Gla residues, which endow them with the capacity to bind to phospholipids.
Initiation of Coagulation. TF exposed at sites of vessel wall injury initiates coagulation via the extrinsic pathway. The small amount of factor VIIa circulating in plasma binds subendothelial TF and the TF-factor VIIa complex, then activates factors X and IX (see Figure 30–2). TF, in the presence of phospholipids and Ca2+, increases the activity of factor VIIa by 30,000-fold.
The intrinsic pathway is initiated in vitro when factor XII, prekallikrein, and high-molecular-weight kininogen interact with kaolin, glass, or another surface to generate small amounts of factor XIIa. Activation of factor XI to factor XIa and factor IX to factor IXa follows. Factor IXa then activates factor X in a reaction accelerated by factor VIIIa, anionic phospholipids, and Ca2+. Optimal thrombin generation depends on the formation of this factor IXa complex because it activates factor X more efficiently than the TF-factor VIIa complex.
Activation of factor XII is not essential for hemostasis, as evidenced by the fact that patients deficient in factor XII, prekallikrein, or high-molecular-weight kininogen do not have excessive bleeding. Factor XI deficiency is associated with a variable and usually mild bleeding disorder.
Fibrinolysis. The pathway of fibrinolysis is summarized in Figure 30–3. The fibrinolytic system dissolves intravascular fibrin through the action of plasmin. To initiate fibrinolysis, plasminogen activators first convert single-chain plasminogen, an inactive precursor, into 2-chain plasmin by cleavage of a specific peptide bond. There are 2 distinct plasminogen activators: tissue plasminogen activator (t-PA) and urokinase plasminogen activator (u-PA), or urokinase. Although both activators are synthesized by endothelial cells, t-PA predominates under most conditions and drives intravascular fibrinolysis, while synthesis of u-PA mainly occurs in response to inflammatory stimuli and promotes extravascular fibrinolysis.
The fibrinolytic system is regulated such that unwanted fibrin thrombi are removed, while fibrin in wounds is preserved to maintain hemostasis. t-PA is released from endothelial cells in response to various stimuli. Released t-PA is rapidly cleared from blood or inhibited by plasminogen activator inhibitor-1 (PAI-1) and, to a lesser extent, plasminogen activator inhibitor-2 (PAI-2); t-PA thus exerts little effect on circulating plasminogen in the absence of fibrin. α2-antiplasmin rapidly inhibits any plasmin that is generated. The catalytic efficiency of t-PA activation of plasminogen increases more than 300-fold in the presence of fibrin, which promotes plasmin generation on its surface.
Plasminogen and plasmin bind to lysine residues on fibrin via 5 loop-like regions near their amino termini, which are known as kringle domains. To inactivate plasmin, α2-antiplasmin binds to the first of these kringle domains and then blocks the active site of plasmin. Because the kringle domains are occupied when plasmin binds to fibrin, plasmin on the fibrin surface is protected from inhibition by α2-antiplasmin and can digest the fibrin. Once the fibrin clot undergoes degradation, α2-antiplasmin rapidly inhibits any plasmin that escapes from this local milieu. To prevent premature clot lysis, factor XIIIa mediates the covalent cross-linking of small amounts of α2-antiplasmin onto fibrin.
When thrombi occlude major arteries or veins, therapeutic doses of plasminogen activators sometimes are administered to degrade the fibrin and rapidly restore blood flow. In high doses, these plasminogen activators promote the generation of so much plasmin that the inhibitory controls are overwhelmed. Plasmin is a relatively nonspecific protease; it also degrades several coagulation factors. Reduction in the levels of these coagulation proteins impairs the capacity for thrombin generation, which can contribute to bleeding. In addition, unopposed plasmin tends to dissolve fibrin in hemostatic plugs as well as that in pathological thrombi, a phenomenon that also increases the risk of bleeding. Therefore, fibrinolytic drugs can be toxic, producing hemorrhage as their major side effect.
Coagulation in vitro. Whole blood normally clots in 4-8 min when placed in a glass tube. Clotting is prevented if a chelating agent such as ethylenediaminetetraacetic acid (EDTA) or citrate is added to bind Ca2+. Recalcified plasma normally clots in 2-4 min. The clotting time after recalcification is shortened to 26-33 seconds by the addition of negatively charged phospholipids and a particulate substance, such as kaolin (aluminum silicate) or celite (diatomaceous earth), which activates factor XII; the measurement of this is termed the activated partial thromboplastin time (aPTT). Alternatively, recalcified plasma clots in 12-14 seconds after addition of “thromboplastin” (a mixture of TF and phospholipids); the measurement of this is termed the prothrombin time (PT).
Natural Anticoagulant Mechanisms. Platelet activation and coagulation do not normally occur within an intact blood vessel. Thrombosis is prevented by several regulatory mechanisms that require a healthy vascular endothelium. NO and PGI2, synthesized by endothelial cells, inhibit platelet activation (see Chapter 33).
Antithrombin is a plasma protein that inhibits coagulation enzymes of the intrinsic and common pathways. Heparan sulfate proteoglycans synthesized by endothelial cells enhance the activity of antithrombin by m1000-fold. Another regulatory system involves protein C, a plasma zymogen that is homologous to factors II, VII, IX, and X; its activity depends on the binding of Ca2+ to Gla residues within its amino-terminal domain. Protein C binds to endothelial protein C receptor (EPCR), which presents protein C to the thrombin–thrombomodulin complex for activation. Activated protein C then dissociates from EPCR and, in combination with protein S, its nonenzymatic Gla-containing cofactor, activated protein C degrades factors Va and VIIIa. Without these activated cofactors, the rates of activation of prothrombin and factor X are greatly diminished. Deficiency of protein C or protein S is associated with an increased risk of pathological thrombus formation.
Tissue factor pathway inhibitor (TFPI), is a natural anticoagulant found in the lipoprotein fraction of plasma. TFPI first binds and inhibits factor Xa, and this binary complex then inhibits factor VIIa. By this mechanism, factor Xa regulates its own generation.
PARENTERAL ANTICOAGULANTS
HEPARIN AND ITS DERIVATIVES
Heparin, a glycosaminoglycan found in the secretory granules of mast cells, is synthesized from UDP-sugar precursors as a polymer of alternating D-glucuronic acid and N-acetyl-D-glucosamine residues.
Heparin is commonly extracted from porcine intestinal mucosa, which is rich in mast cells, and preparations may contain small amounts of other glycosaminoglycans. The biological activities among different commercial preparations of heparin are similar (~150 USP units/mg). A USP unit reflects the quantity of heparin that prevents 1 mL of citrated sheep plasma from clotting for 1 h after the addition of 0.2 mL of 1% CaCl2. European manufacturers measure potency with an antifactor Xa assay. To determine heparin potency, residual factor Xa activity in the sample is compared with that detected in controls containing known concentrations of an international heparin standard. When assessed this way, heparin potency is expressed in international units per mg. Effective October 1, 2009, the new USP unit dose has been harmonized to the international unit dose. As a result, the new USP unit dose is less potent than the old USP unit dose by ~10%, and heparin doses using the new USP units will have to increase slightly to achieve the same level of anticoagulation.
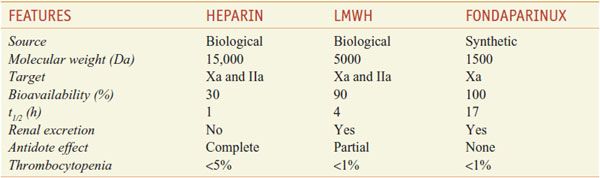
HEPARIN DERIVATIVES. Derivatives of heparin in current use include low-molecular-weight heparins (LMWHs) and fondaparinux (see their comparison in Table 30–1).
Table 30–1
Comparison of Heparin, LMWH, and Fondaparinux
Mechanism of Action. Heparin, LMWHs, and fondaparinux have no intrinsic anticoagulant activity; rather, these agents bind to antithrombin and accelerate the rate at which it inhibits various coagulation proteases. Synthesized in the liver, antithrombin circulates in plasma at an approximate concentration of 2.6 μM. Antithrombin inhibits activated coagulation factors involved in the intrinsic and common pathways but has relatively little activity against factor VIIa. Antithrombin is a “suicide substrate” for these proteases; inhibition occurs when the protease attacks a specific Arg–Ser peptide bond in the reactive center loop of antithrombin and becomes trapped as a stable 1:1 complex. Heparin binds to antithrombin via a specific pentasaccharide sequence that contains a 3-O-sulfated glucosamine residue (Figure 30–4). Pentasaccharide binding to antithrombin induces a conformational change in antithrombin that renders its reactive site more accessible to the target protease (Figure 30–5). This conformational change accelerates the rate of factor Xa inhibition by at least 2 orders of magnitude but has no effect on the rate of thrombin inhibition. To enhance the rate of thrombin inhibition by antithrombin, heparin serves as a catalytic template to which both the inhibitor and the protease bind. Only heparin molecules composed of 18 or more saccharide units (molecular weight >5400 Da) are of sufficient length to bridge antithrombin and thrombin together. Consequently, by definition, heparin catalyzes the rates of factor Xa and thrombin to a similar extent, as expressed by an antifactor Xa to antifactor IIa (thrombin) ratio of 1:1. In contrast, at least half of LMWH molecules (mean molecular weight of 5000 Da, ~17 saccharide units) are too short to provide this bridging function and have no effect on the rate of thrombin inhibition by antithrombin. Because these shorter molecules still induce the conformational change in antithrombin that accelerates inhibition of factor Xa, LMWHs have greater antifactor Xa activity than antifactor IIa activity, and the ratio ranges from 3:1 to 2:1 depending on the preparation. Fondaparinux, an analog of the pentasaccharide sequence in heparin or LMWHs that mediates their interaction with antithrombin, has only antifactor Xa activity because it is too short to bridge antithrombin to thrombin (see Figure 30–5).
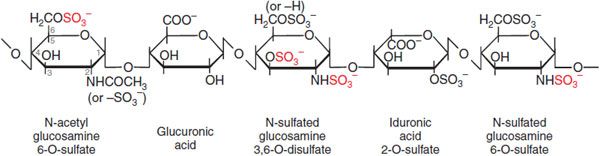
Figure 30–4 The antithrombin-binding pentasaccharide structure of heparin. Sulfate groups required for binding to antithrombin are indicated in red.
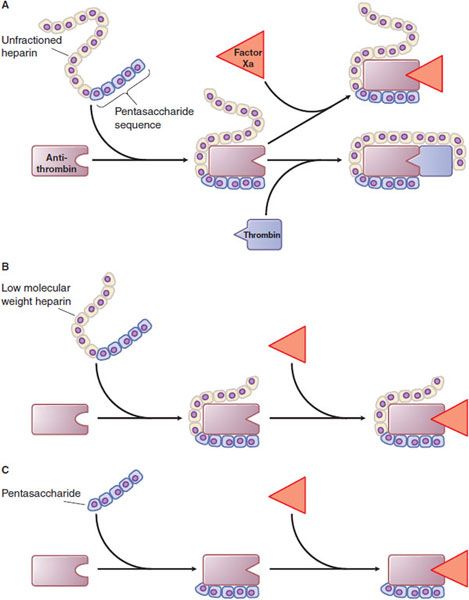
Figure 30–5 Mechanism of action of heparin, low-molecular-weight heparin (LMWH), and fondaparinux, a synthetic pentasaccharide. A. Heparin binds to antithrombin via its pentasaccharide sequence. This induces a conformational change in the reactive center loop of antithrombin that accelerates its interaction with factor Xa. To potentiate thrombin inhibition, heparin must simultaneously bind to antithrombin and thrombin. Only heparin chains composed of at least 18 saccharide units (MW ~5,400 Da) are of sufficient length to perform this bridging function. With a mean MW ~15,000 Da, virtually all of the heparin chains are long enough to do this. B. LMWH has greater capacity to potentiate factor Xa inhibition by antithrombin than thrombin because at least half of the LMWH chains (mean MW ~4,500-5,000 Da) are too short to bridge antithrombin to thrombin. C. The pentasaccharide accelerates only factor Xa inhibition by antithrombin; the pentasaccharide is too short to bridge antithrombin to thrombin.
Heparin, LMWHs, and fondaparinux act in a catalytic fashion. After binding to antithrombin and promoting the formation of covalent complexes between antithrombin and target proteases, the heparin, LMWH, or fondaparinux dissociates from the complex and can then catalyze other antithrombin molecules.
Platelet factor 4, a cationic protein released from the α granules during platelet activation, binds heparin and prevents it from interacting with antithrombin. This phenomenon may limit the activity of heparin in the vicinity of platelet-rich thrombi. Because LMWH and fondaparinux have a lower affinity for platelet factor 4, these agents may retain their activity in the vicinity of such thrombi to a greater extent than heparin.
Miscellaneous Pharmacological Effects. High doses of heparin can interfere with platelet aggregation and prolong bleeding time. In contrast, LMWHs and fondaparinux have little effect on platelets. Heparin “clears” lipemic plasma in vivo by causing the release of lipoprotein lipase into the circulation. Lipoprotein lipase hydrolyzes triglycerides to glycerol and free fatty acids. The clearing of lipemic plasma may occur at concentrations of heparin below those necessary to produce an anticoagulant effect.
Clinical Use. Heparin, LMWH, and fondaparinux can be used to initiate treatment of venous thrombosis and pulmonary embolism because of their rapid onset of action. An oral vitamin K antagonist, such as warfarin, usually is started concurrently, and the heparin or heparin derivative is continued for at least 5 days to allow warfarin to achieve its full therapeutic effect. Heparin, LMWH, or fondaparinux also can be used in the initial management of patients with unstable angina or acute myocardial infarction. For most of these indications, LMWHs and fondaparinux have replaced continuous heparin infusions because of their pharmacokinetic advantages, which permit subcutaneous administration once or twice daily in fixed or weight-adjusted doses without coagulation monitoring. Thus, LMWHs or fondaparinux can be used for out-of-hospital management of patients with venous thrombosis or pulmonary embolism.
Heparin and LMWH are used during coronary balloon angioplasty with or without stent placement to prevent thrombosis. Fondaparinux is not used in this setting because of the risk of catheter thrombosis, a complication caused by catheter-induced activation of factor XII; longer heparin molecules are better than shorter ones for blocking this process. Cardiopulmonary bypass circuits also activate factor XII, which can cause clotting of the oxygenator. Heparin remains the agent of choice for surgery requiring cardiopulmonary bypass. Heparin also is used to treat selected patients with disseminated intravascular coagulation. Subcutaneous administration of low-dose heparin remains the recommended regimen for the prevention of postoperative deep venous thrombosis (DVT) and pulmonary embolism in patients undergoing major abdominothoracic surgery or who are at risk of developing thromboembolic disease.
Unlike warfarin, heparin, LMWH, and fondaparinux do not cross the placenta and have not been associated with fetal malformations, making them the drugs of choice for anticoagulation during pregnancy. Heparin, LMWH, and fondaparinux do not appear to increase fetal mortality or prematurity. If possible, the drugs should be discontinued 24 h before delivery to minimize the risk of postpartum bleeding.
ADME. Heparin, LMWHs, and fondaparinux are not absorbed through the GI mucosa and must be given parenterally. Heparin is given by continuous intravenous infusion, intermittent infusion every 4-6 h, or subcutaneous injection every 8-12 h. Heparin has an immediate onset of action when given intravenously. In contrast, there is considerable variation in the bioavailability of heparin given subcutaneously, and the onset of action is delayed 1-2 h. LMWH and fondaparinux are absorbed more uniformly after subcutaneous injection. The t1/2 of heparin in plasma depends on the dose administered. When doses of 100, 400, or 800 units/kg of heparin are injected intravenously, the half-lives of the anticoagulant activities are ~1, 2.5, and 5 h. Heparin appears to be cleared and degraded primarily by the reticuloendothelial system; a small amount of undegraded heparin appears in the urine.
LMWHs and fondaparinux have longer biological half-lives than heparin, 4-6 h and L17 h, respectively. Because these smaller heparin fragments are cleared almost exclusively by the kidneys, the drugs can accumulate in patients with renal impairment and lead to bleeding. Both LMWH and fondaparinux are contraindicated in patients with a creatinine clearance <30 mL/min. In addition, fondaparinux is contraindicated in patients with body weight <50 kg undergoing hip fracture, hip replacement, knee replacement surgery, or abdominal surgery.
Administration and Monitoring. Full-dose heparin therapy usually is administered by continuous intravenous infusion. Treatment of venous thromboembolism is initiated with a fixed-dose bolus injection of 5000 units or with a weight-adjusted bolus, followed by 800-1600 units/h delivered by an infusion pump. Therapy routinely is monitored by measuring the aPTT. The therapeutic range for heparin is considered to be that which is equivalent to a plasma heparin level of 0.3-0.7 units/mL, as determined with an antifactor Xa assay. An aPTT 2-3 times the normal mean aPTT value generally is assumed to be therapeutic. The aPTT should be measured initially and the infusion rate adjusted every 6 h. Once a steady dosage schedule has been established in a stable patient, daily laboratory monitoring usually is sufficient. Very high doses of heparin are required to prevent coagulation during cardiopulmonary bypass. The aPTT is infinitely prolonged over the dosage range used. A less sensitive coagulation test, such as the activated clotting time, is employed to monitor therapy in this situation.
For therapeutic purposes, heparin also can be administered subcutaneously on a twice-daily basis. A total daily dose of ~35,000 units administered as divided doses every 8-12 h usually is sufficient to achieve an aPTT of twice the control value (measured midway between doses). For low-dose heparin therapy (to prevent DVT and thromboembolism in hospitalized medical or surgical patients), a subcutaneous dose of 5000 units is given 2-3 times daily.
LMWH PREPARATIONS include Enoxaparin (LOVENOX), dalteparin (FRAGMIN), tinzaparin (INNOHEP, others), ardeparin (NORMIFLO), nadroparin (FRAXIPARINE, others), and reviparin (CLIVARINE) (the latter 3 are not currently available in the United States). These agents differ considerably; do not assume that 2 preparations that have similar antifactor Xa activity will have equivalent antithrombotic effects. Because LMWHs produce a relatively predictable anticoagulant response, monitoring is not done routinely. Patients with renal impairment may require monitoring with an antifactor Xa assay because this condition may prolong the t1/2 and slow the elimination of LMWHs. Obese patients and children given LMWHs also may require monitoring.
FONDAPARINUX (ARIXTRA) is administered by subcutaneous injection, reaches peak plasma levels in 2 h, and is excreted in the urine (t1/2 17 h). It should not be used in patients with renal failure. Fondaparinux can be given once a day at a fixed dose without coagulation monitoring. Fondaparinux appears to be much less likely than heparin or LMWH to trigger the syndrome of heparin-induced thrombocytopenia. Fondaparinux is approved for thromboprophylaxis in patients undergoing hip or knee surgery or surgery for hip fracture, and for initial therapy in patients with pulmonary embolism or DVT.
IDRAPARINUX, a hypermethylated version of fondaparinux with a t1/2 of 80 h; it is given subcutaneously once-weekly. To overcome the lack of an antidote, a biotin moiety was added to idraparinux to generate idrabiotaparinux, which can be neutralized with intravenous avidin. Ongoing phase III clinical trials are comparing idrabiotaparinux with warfarin for treatment of pulmonary embolism or for stroke prevention in patients with atrial fibrillation. Idraparinux, idrabiotaparinux, and avidin are not available for routine clinical use.
Heparin Resistance.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


