FIGURE 411-1 Pubertal events in males. Sexual maturity ratings for genitalia and pubic hair and divided into five stages. (From WA Marshall, JM Tanner: Variations in the pattern of pubertal changes in boys. Arch Dis Child 45:13, 1970.)
The early stages of puberty are characterized by nocturnal surges of LH and FSH. Growth of the testes is usually the first sign of puberty, reflecting an increase in seminiferous tubule volume. Increasing levels of testosterone deepen the voice and increase muscle growth. Conversion of testosterone to DHT leads to growth of the external genitalia and pubic hair. DHT also stimulates prostate and facial hair growth and initiates recession of the temporal hairline. The growth spurt occurs at a testicular volume of about 10–12 mL. GH increases early in puberty and is stimulated in part by the rise in gonadal steroids. GH increases the level of insulin-like growth factor I (IGF-I), which enhances linear bone growth. The prolonged pubertal exposure to gonadal steroids (mainly estradiol) ultimately causes epiphyseal closure and limits further bone growth.
REGULATION OF TESTICULAR FUNCTION
REGULATION OF THE HYPOTHALAMIC-PITUITARY-TESTIS AXIS IN ADULT MAN
Hypothalamic GnRH regulates the production of the pituitary gonadotropins LH and FSH (Fig. 411-2). GnRH is released in discrete pulses approximately every 2 h, resulting in corresponding pulses of LH and FSH. These dynamic hormone pulses account in part for the wide variations in LH and testosterone, even within the same individual. LH acts primarily on the Leydig cell to stimulate testosterone synthesis. The regulatory control of androgen synthesis is mediated by testosterone and estrogen feedback on both the hypothalamus and the pituitary. FSH acts on the Sertoli cell to regulate spermatogenesis and the production of Sertoli products such as inhibin B, which acts to selectively suppress pituitary FSH. Despite these somewhat distinct Leydig and Sertoli cell–regulated pathways, testis function is integrated at several levels: GnRH regulates both gonadotropins; spermatogenesis requires high levels of testosterone; and numerous paracrine interactions between Leydig and Sertoli cells are necessary for normal testis function.
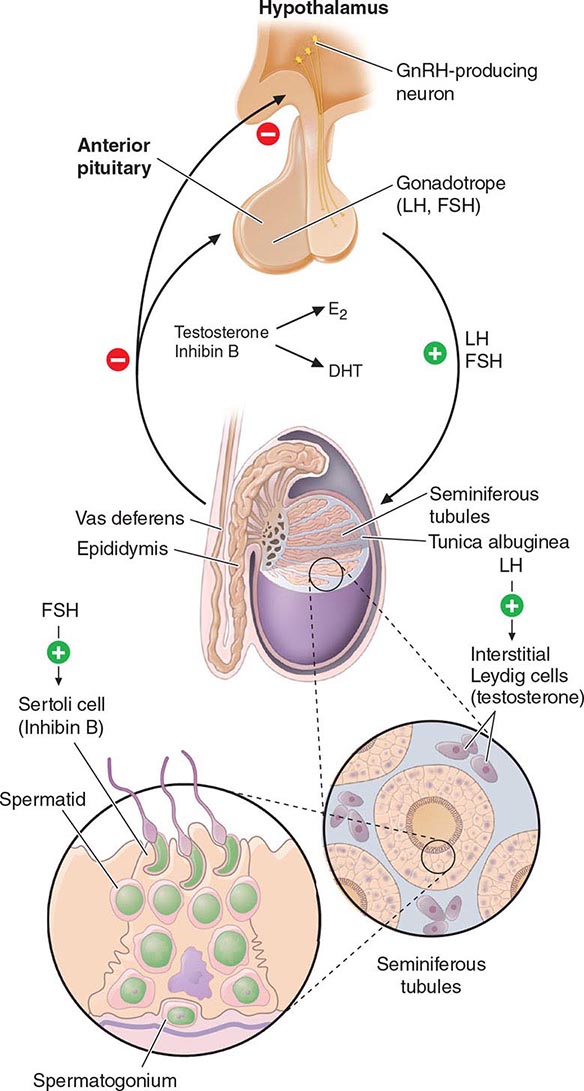
FIGURE 411-2 Human pituitary gonadotropin axis, structure of testis, and seminiferous tubule. E2, 17β-estradiol; DHT, dihydrotestosterone; FSH, follicle-stimulating hormones; GnRH, gonadotropin-releasing; LH, luteinizing hormone.
THE LEYDIG CELL: ANDROGEN SYNTHESIS
LH binds to its seven-transmembrane, G protein–coupled receptor to activate the cyclic AMP pathway. Stimulation of the LH receptor induces steroid acute regulatory (StAR) protein, along with several steroidogenic enzymes involved in androgen synthesis. LH receptor mutations cause Leydig cell hypoplasia or agenesis, underscoring the importance of this pathway for Leydig cell development and function. The rate-limiting process in testosterone synthesis is the delivery of cholesterol by the StAR protein to the inner mitochondrial membrane. Peripheral benzodiazepine receptor, a mitochondrial cholesterol-binding protein, is also an acute regulator of Leydig cell steroidogenesis. The five major enzymatic steps involved in testosterone synthesis are summarized in Fig. 411-3. After cholesterol transport into the mitochondrion, the formation of pregnenolone by CYP11A1 (side chain cleavage enzyme) is a limiting enzymatic step. The 17α-hydroxylase and the 17,20-lyase reactions are catalyzed by a single enzyme, CYP17; posttranslational modification (phosphorylation) of this enzyme and the presence of specific enzyme cofactors confer 17,20-lyase activity selectively in the testis and zona reticularis of the adrenal gland. Testosterone can be converted to the more potent DHT by 5α-reductase, or it can be aromatized to estradiol by CYP19 (aromatase). Two isoforms of steroid 5α-reductase, SRD5A1 and SRD5A2, have been described; all known kindreds with 5α-reductase deficiency have had mutations in SRD5A2, the predominant form in the prostate and the skin.
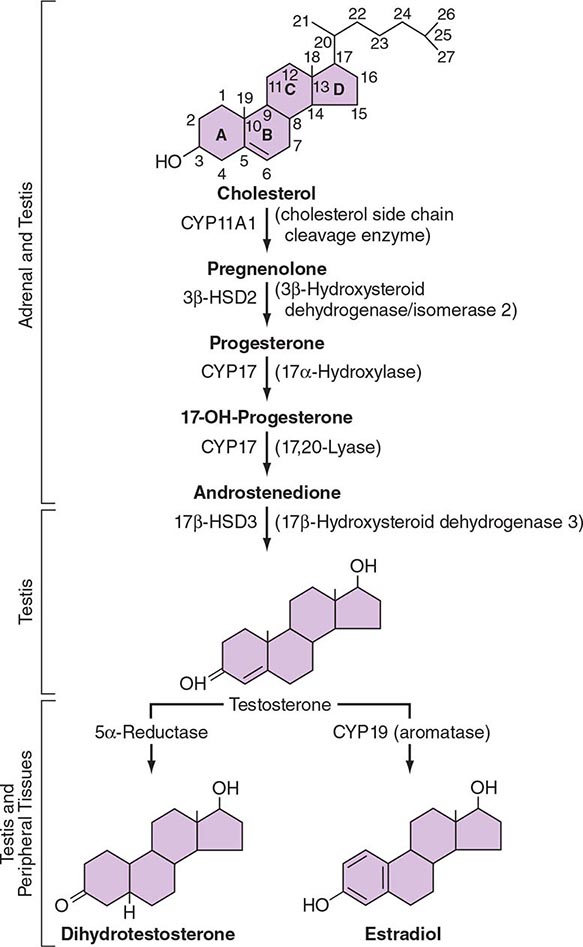
FIGURE 411-3 The biochemical pathway in the conversion of 27-carbon sterol cholesterol to androgens and estrogens.
Testosterone Transport and Metabolism In males, 95% of circulating testosterone is derived from testicular production (3–10 mg/d). Direct secretion of testosterone by the adrenal and the peripheral conversion of androstenedione to testosterone collectively account for another 0.5 mg/d of testosterone. Only a small amount of DHT (70 μg/d) is secreted directly by the testis; most circulating DHT is derived from peripheral conversion of testosterone. Most of the daily production of estradiol (~45 μg/d) in men is derived from aromatase-mediated peripheral conversion of testosterone and androstenedione.
Circulating testosterone is bound to two plasma proteins: sex hormone–binding globulin (SHBG) and albumin (Fig. 411-4). SHBG binds testosterone with much greater affinity than albumin. Only 0.5–3% of testosterone is unbound. According to the “free hormone” hypothesis, only the unbound fraction is biologically active; however, albumin-bound hormone dissociates readily in the capillaries and may be bioavailable. SHBG-bound testosterone also may be internalized through endocytic pits by binding to a protein called megalin. SHBG concentrations are decreased by androgens, obesity, diabetes mellitus, insulin, and nephrotic syndrome. Conversely, estrogen administration, hyperthyroidism, many chronic inflammatory illnesses, infections such as HIV or hepatitis B and C, and aging are associated with high SHBG concentrations.
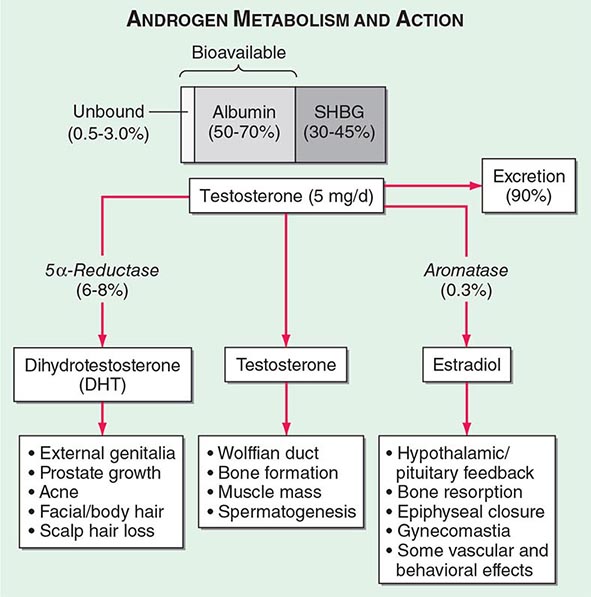
FIGURE 411-4 Androgen metabolism and actions. SHBG, sex hormone–binding globulin.
Testosterone is metabolized predominantly in the liver, although some degradation occurs in peripheral tissues, particularly the prostate and the skin. In the liver, testosterone is converted by a series of enzymatic steps that involve 5α- and 5β-reductases, 3α- and 3β-hydroxysteroid dehydrogenases, and 17β-hydroxysteroid dehydrogenase into androsterone, etiocholanolone, DHT, and 3-α-androstanediol. These compounds undergo glucuronidation or sulfation before being excreted by the kidneys.
Mechanism of Androgen Action Testosterone exerts some of its biologic effects by binding to androgen receptor, either directly or after its conversion to DHT by the steroid 5-α reductase. Testosterone’s effects on the skeletal muscle, erythropoiesis, and bone in men do not require its obligatory conversion to DHT. However, the conversion of testosterone to DHT is necessary for the masculinization of the urogenital sinus and genital tubercle. Aromatization of testosterone to estradiol mediates additional effects of testosterone on the bone resorption, epiphyseal closure, sexual desire, vascular endothelium, and fat. DHT can also be converted in some tissues by 3-keto reductase/3β-hydroxysteroid dehydrogenase enzymes to 5α-androstane-3β,17β-diol, which is a high-affinity ligand and agonist of estrogen receptor β.
The androgen receptor (AR) is structurally related to the nuclear receptors for estrogen, glucocorticoids, and progesterone (Chap. 400e). The AR is encoded by a gene on the long arm of the × chromosome and has a molecular mass of about 110 kDa. A polymorphic region in the amino terminus of the receptor, which contains a variable number of glutamine repeats, modifies the transcriptional activity of the receptor. The AR protein is distributed in both the cytoplasm and the nucleus. The ligand binding to the AR induces conformational changes that allow the recruitment and assembly of tissue-specific cofactors and causes it to translocate into the nucleus, where it binds to DNA or other transcription factors already bound to DNA. Thus, the AR is a ligand-regulated transcription factor that regulates the expression of androgen-dependent genes in a tissue-specific manner. Some androgen effects may be mediated by nongenomic AR signal transduction pathways. Testosterone binds to AR with half the affinity of DHT. The DHT-AR complex also has greater thermostability and a slower dissociation rate than the testosterone-AR complex. However, the molecular basis for selective testosterone versus DHT actions remains incompletely explained.
THE SEMINIFEROUS TUBULES: SPERMATOGENESIS
The seminiferous tubules are convoluted, closed loops with both ends emptying into the rete testis, a network of progressively larger efferent ducts that ultimately form the epididymis (Fig. 411-2). The seminiferous tubules total about 600 m in length and comprise about two-thirds of testis volume. The walls of the tubules are formed by polarized Sertoli cells that are apposed to peritubular myoid cells. Tight junctions between Sertoli cells create a blood-testis barrier. Germ cells compose the majority of the seminiferous epithelium (~60%) and are intimately embedded within the cytoplasmic extensions of the Sertoli cells, which function as “nurse cells.” Germ cells progress through characteristic stages of mitotic and meiotic divisions. A pool of type A spermatogonia serve as stem cells capable of self-renewal. Primary spermatocytes are derived from type B spermatogonia and undergo meiosis before progressing to spermatids that undergo spermiogenesis (a differentiation process involving chromatin condensation, acquisition of an acrosome, elongation of cytoplasm, and formation of a tail) and are released from Sertoli cells as mature spermatozoa. The complete differentiation process into mature sperm requires 74 days. Peristaltic-type action by peritubular myoid cells transports sperm into the efferent ducts. The spermatozoa spend an additional 21 days in the epididymis, where they undergo further maturation and capacitation. The normal adult testes produce >100 million sperm per day.
Naturally occurring mutations in the FSHβ gene and in the FSH receptor confirm an important, but not essential, role for this pathway in spermatogenesis. Females with these mutations are hypogonadal and infertile because ovarian follicles do not mature; males exhibit variable degrees of reduced spermatogenesis, presumably because of impaired Sertoli cell function. Because Sertoli cells produce inhibin B, an inhibitor of FSH, seminiferous tubule damage (e.g., by radiation) causes a selective increase of FSH. Testosterone reaches very high concentrations locally in the testis and is essential for spermatogenesis. The cooperative actions of FSH and testosterone are important in the progression of meiosis and spermiation. FSH and testosterone regulate germ cell survival via the intrinsic and the extrinsic apoptotic mechanisms. FSH may also play an important role in supporting spermatogonia. Gonadotropin-regulated testicular RNA helicase (GRTH/DDX25), a testis-specific gonadotropin/androgen-regulated RNA helicase, is present in germ cells and Leydig cells and may be an important factor in the paracrine regulation of germ cell development. Several cytokines and growth factors are also involved in the regulation of spermatogenesis by paracrine and autocrine mechanisms. A number of knockout mouse models exhibit impaired germ cell development or spermatogenesis, presaging possible mutations associated with male infertility. The human Y chromosome contains a small pseudoautosomal region that can recombine with homologous regions of the × chromosome. Most of the Y chromosome does not recombine with the × chromosome and is referred to as the male-specific region of the Y (MSY). The MSY contains 156 transcription units that encode for 26 proteins, including nine families of Y-specific multicopy genes; many of these Y-specific genes are also testis-specific and necessary for spermatogenesis. Microdeletions of several Y chromosome azoospermia factor (AZF) genes (e.g., RNA-binding motif, RBM; deleted in azoospermia, DAZ) are associated with oligospermia or azoospermia.
TREATMENT | MALE FACTOR INFERTILITY |
Treatment options for male factor infertility have expanded greatly in recent years. Secondary hypogonadism is highly amenable to treatment with pulsatile GnRH or gonadotropins (see below). Assisted reproductive technologies such as the in vitro fertilization (IVF) and intracytoplasmic sperm injection (ICSI) have provided new opportunities for patients with primary testicular failure and disorders of sperm transport. Choice of initial treatment options depends on sperm concentration and motility. Expectant management should be attempted initially in men with mild male factor infertility (sperm count of 15–20 × 106/mL and normal motility). Moderate male factor infertility (10–15 × 106/mL and 20–40% motility) should begin with intrauterine insemination alone or in combination with treatment of the female partner with clomiphene or gonadotropins, but it may require IVF with or without ICSI. For men with a severe defect (sperm count of <10 × 106/mL, 10% motility), IVF with ICSI or donor sperm should be used.
CLINICAL AND LABORATORY EVALUATION OF MALE REPRODUCTIVE FUNCTION
HISTORY AND PHYSICAL EXAMINATION
The history should focus on developmental stages such as puberty and growth spurts, as well as androgen-dependent events such as early morning erections, frequency and intensity of sexual thoughts, and frequency of masturbation or intercourse. Although libido and the overall frequency of sexual acts are decreased in androgen-deficient men, young hypogonadal men may achieve erections in response to visual erotic stimuli. Men with acquired androgen deficiency often report decreased energy and increased irritability.
The physical examination should focus on secondary sex characteristics such as hair growth, gynecomastia, testicular volume, prostate, and height and body proportions. Eunuchoid proportions are defined as an arm span >2 cm greater than height and suggest that androgen deficiency occurred before epiphyseal fusion. Hair growth in the face, axilla, chest, and pubic regions is androgen-dependent; however, changes may not be noticeable unless androgen deficiency is severe and prolonged. Ethnicity also influences the intensity of hair growth (Chap. 68). Testicular volume is best assessed by using a Prader orchidometer. Testes range from 3.5 to 5.5 cm in length, which corresponds to a volume of 12–25 mL. Advanced age does not influence testicular size, although the consistency becomes less firm. Asian men generally have smaller testes than Western Europeans, independent of differences in body size. Because of its possible role in infertility, the presence of varicocele should be sought by palpation while the patient is standing; it is more common on the left side. Patients with Klinefelter’s syndrome have markedly reduced testicular volumes (1–2 mL). In congenital hypogonadotropic hypogonadism, testicular volumes provide a good index for the degree of gonadotropin deficiency and the likelihood of response to therapy.
GONADOTROPIN AND INHIBIN MEASUREMENTS
LH and FSH are measured using two-site immunoradiometric, immunofluorometric, or chemiluminescent assays, which have very low cross-reactivity with other pituitary glycoprotein hormones and human chorionic gonadotropin (hCG) and have sufficient sensitivity to measure the low levels present in patients with hypogonadotropic hypogonadism. In men with a low testosterone level, an LH level can distinguish primary (high LH) versus secondary (low or inappropriately normal LH) hypogonadism. An elevated LH level indicates a primary defect at the testicular level, whereas a low or inappropriately normal LH level suggests a defect at the hypothalamic-pituitary level. LH pulses occur about every 1–3 h in normal men. Thus, gonadotropin levels fluctuate, and samples should be pooled or repeated when results are equivocal. FSH is less pulsatile than LH because it has a longer half-life. Selective increase in FSH suggests damage to the seminiferous tubules. Inhibin B, a Sertoli cell product that suppresses FSH, is reduced with seminiferous tubule damage. Inhibin B is a dimer with α-βB subunits and is measured by two-site immunoassays.
GnRH Stimulation Testing The GnRH test is performed by measuring LH and FSH concentrations at baseline and at 30 and 60 min after intravenous administration of 100 μg of GnRH. A minimally acceptable response is a twofold LH increase and a 50% FSH increase. In the prepubertal period or with severe GnRH deficiency, the gonadotrope may not respond to a single bolus of GnRH because it has not been primed by endogenous hypothalamic GnRH; in these patients, GnRH responsiveness may be restored by chronic, pulsatile GnRH administration. With the availability of sensitive and specific LH assays, GnRH stimulation testing is used rarely except to evaluate gonadotrope function in patients who have undergone pituitary surgery or have a space-occupying lesion in the hypothalamic-pituitary region.
TESTOSTERONE ASSAYS
Total Testosterone Total testosterone includes both unbound and protein-bound testosterone and is measured by radioimmunoassays, immunometric assays, or liquid chromatography tandem mass spectrometry (LC-MS/MS). LC-MS/MS involves extraction of serum by organic solvents, separation of testosterone from other steroids by high-performance liquid chromatography and mass spectrometry, and quantitation of unique testosterone fragments by mass spectrometry. LC-MS/MS provides accurate and sensitive measurements of testosterone levels even in the low range and is emerging as the method of choice for testosterone measurement. Laboratories that have been certified by the Centers for Disease Control and Prevention (CDC) Hormone Standardization Program for Testosterone (HoST) can ensure that testosterone measurements are accurate and calibrated to an international standard. A single fasting morning sample provides a good approximation of the average testosterone concentration with the realization that testosterone levels fluctuate in response to pulsatile LH. Testosterone is generally lower in the late afternoon and is reduced by acute illness. The testosterone concentration in healthy young men ranges from 300 to 1000 ng/dL in most laboratories, and efforts are under way to generate harmonized population-based reference ranges that can be applied to all CDC-certified laboratories. Alterations in SHBG levels due to aging, obesity, diabetes mellitus, hyperthyroidism, some types of medications, or chronic illness or on a congenital basis can affect total testosterone levels. Heritable factors contribute substantially to the population-level variation in testosterone levels, and genome-wide association studies have revealed polymorphisms in the SHBG gene as important contributors to variation in testosterone levels.
Measurement of Unbound Testosterone Levels Most circulating testosterone is bound to SHBG and to albumin; only 0.5–3% of circulating testosterone is unbound, or “free.” The unbound testosterone concentration can be measured by equilibrium dialysis or calculated from total testosterone, SHBG, and albumin concentrations. Recent research has shown that testosterone binding to SHBG is a multistep process that involves complex homoallostery within the SHBG dimer; a novel allosteric model of testosterone binding to SHBG dimers provides good estimates of free testosterone concentrations. The previous law of mass action equations based on linear models of testosterone binding to SHBG have been shown to be erroneous. Tracer analogue methods are relatively inexpensive and convenient, but they are inaccurate. Bioavailable testosterone refers to unbound testosterone plus testosterone that is loosely bound to albumin; it can be determined by the ammonium sulfate precipitation method.
hCG Stimulation Test The hCG stimulation test is performed by administering a single injection of 1500–4000 IU of hCG intramuscularly and measuring testosterone levels at baseline and 24, 48, 72, and 120 h after hCG injection. An alternative regimen involves three injections of 1500 units of hCG on successive days and measuring testosterone levels 24 h after the last dose. An acceptable response to hCG is a doubling of the testosterone concentration in adult men. In prepubertal boys, an increase in testosterone to >150 ng/dL indicates the presence of testicular tissue. No response may indicate an absence of testicular tissue or marked impairment of Leydig cell function. Measurement of MIS, a Sertoli cell product, is also used to detect the presence of testes in prepubertal boys with cryptorchidism.
SEMEN ANALYSIS
Semen analysis is the most important step in the evaluation of male infertility. Samples are collected by masturbation following a period of abstinence for 2–3 days. Semen volumes and sperm concentrations vary considerably among fertile men, and several samples may be needed before concluding that the results are abnormal. Analysis should be performed within an hour of collection. Using semen samples from over 4500 men in 14 countries, whose partners had a time-to-pregnancy of less than 12 months, the World Health Organization (WHO) has generated the following one-sided reference limits for semen parameters: semen volume, 1.5 mL; total sperm number, 39 million per ejaculate; sperm concentration, 15 million/mL; vitality, 58% live; progressive motility, 32%; total (progressive + nonprogressive) motility, 40%; morphologically normal forms, 4.0%. Some men with low sperm counts are nevertheless fertile. A variety of tests for sperm function can be performed in specialized laboratories, but these add relatively little to the treatment options.
TESTICULAR BIOPSY
Testicular biopsy is useful in some patients with oligospermia or azoospermia as an aid in diagnosis and indication for the feasibility of treatment. Using local anesthesia, fine-needle aspiration biopsy is performed to aspirate tissue for histology. Alternatively, open biopsies can be performed under local or general anesthesia when more tissue is required. A normal biopsy in an azoospermic man with a normal FSH level suggests obstruction of the vas deferens, which may be correctable surgically. Biopsies are also used to harvest sperm for ICSI and to classify disorders such as hypospermatogenesis (all stages present but in reduced numbers), germ cell arrest (usually at primary spermatocyte stage), and Sertoli cell–only syndrome (absent germ cells) or hyalinization (sclerosis with absent cellular elements).
DISORDERS OF SEXUAL DIFFERENTIATION
See Chap. 410.
DISORDERS OF PUBERTY
The onset and tempo of puberty varies greatly in the general population and is affected by genetic and environmental factors. Although some of the variance in the timing of puberty is explained by heritable factors, the genes involved remain unknown.
PRECOCIOUS PUBERTY
Puberty in boys before age 9 is considered precocious. Isosexual precocity refers to premature sexual development consistent with phenotypic sex and includes features such as the development of facial hair and phallic growth. Isosexual precocity is divided into gonadotropin-dependent and gonadotropin-independent causes of androgen excess (Table 411-1). Heterosexual precocity refers to the premature development of estrogenic features in boys, such as breast development.
CAUSES OF PRECOCIOUS OR DELAYED PUBERTY IN BOYS |
Abbreviations: CNS, central nervous system; GnRH, gonadotropin-releasing hormone; hCG, human chronic gonadotropin; LH, luteinizing hormone.
Gonadotropin-Dependent Precocious Puberty This disorder, called central precocious puberty (CPP), is less common in boys than in girls. It is caused by premature activation of the GnRH pulse generator, sometimes because of central nervous system (CNS) lesions such as hypothalamic hamartomas, but it is often idiopathic. CPP is characterized by gonadotropin levels that are inappropriately elevated for age. Because pituitary priming has occurred, GnRH elicits LH and FSH responses typical of those seen in puberty or in adults. Magnetic resonance imaging (MRI) should be performed to exclude a mass, structural defect, infection, or inflammatory process. Mutations in MKRN3, an imprinted gene encoding makorin ring-finger protein 3, which is expressed only from the paternally inherited allele, have been associated with CPP.
Gonadotropin-Independent Precocious Puberty In gonadotropin-independent precocious puberty, androgens from the testis or the adrenal are increased, but gonadotropins are low. This group of disorders includes hCG-secreting tumors; congenital adrenal hyperplasia; sex steroid–producing tumors of the testis, adrenal, and ovary; accidental or deliberate exogenous sex steroid administration; hypothyroidism; and activating mutations of the LH receptor or Gsα subunit.
FAMILIAL MALE-LIMITED PRECOCIOUS PUBERTY Also called testotoxicosis, familial male-limited precocious puberty is an autosomal dominant disorder caused by activating mutations in the LH receptor, leading to constitutive stimulation of the cyclic AMP pathway and testosterone production. Clinical features include premature androgenization in boys, growth acceleration in early childhood, and advanced bone age followed by premature epiphyseal fusion. Testosterone is elevated, and LH is suppressed. Treatment options include inhibitors of testosterone synthesis (e.g., ketoconazole), AR antagonists (e.g., flutamide and bicalutamide), and aromatase inhibitors (e.g., anastrazole).
MCCUNE-ALBRIGHT SYNDROME This is a sporadic disorder caused by somatic (postzygotic) activating mutations in the Gsα subunit that links G protein–coupled receptors to intracellular signaling pathways (Chap. 426e). The mutations impair the guanosine triphosphatase activity of the Gsα protein, leading to constitutive activation of adenylyl cyclase. Like activating LH receptor mutations, this stimulates testosterone production and causes gonadotropin-independent precocious puberty. In addition to sexual precocity, affected individuals may have autonomy in the adrenals, pituitary, and thyroid glands. Café au lait spots are characteristic skin lesions that reflect the onset of the somatic mutations in melanocytes during embryonic development. Polyostotic fibrous dysplasia is caused by activation of the parathyroid hormone receptor pathway in bone. Treatment is similar to that in patients with activating LH receptor mutations. Bisphosphonates have been used to treat bone lesions.
CONGENITAL ADRENAL HYPERPLASIA Boys with congenital adrenal hyperplasia (CAH) who are not well controlled with glucocorticoid suppression of adrenocorticotropic hormone (ACTH) can develop premature virilization because of excessive androgen production by the adrenal gland (Chaps. 406 and 410). LH is low, and the testes are small. Adrenal rests may develop within the testis of poorly controlled patients with CAH because of chronic ACTH stimulation; adrenal rests do not require surgical removal and regress with effective glucocorticoid therapy. Some children with CAH may develop gonadotropin-dependent precocious puberty with early maturation of the hypothalamic-pituitary-gonadal axis, elevated gonadotropins, and testicular growth.
Heterosexual Sexual Precocity Breast enlargement in prepubertal boys can result from familial aromatase excess, estrogen-producing tumors in the adrenal gland, Sertoli cell tumors in the testis, marijuana smoking, or exogenous estrogens or androgens. Occasionally, germ cell tumors that secrete hCG can be associated with breast enlargement due to excessive stimulation of estrogen production (see “Gynecomastia,” below).
TREATMENT | PRECOCIOUS PUBERTY |
In patients with a known cause (e.g., a CNS lesion or a testicular tumor), therapy should be directed toward the underlying disorder. In patients with idiopathic CPP, long-acting GnRH analogues can be used to suppress gonadotropins and decrease testosterone, halt early pubertal development, delay accelerated bone maturation, prevent early epiphyseal closure, promote final height gain, and mitigate the psychosocial consequences of early pubertal development without causing osteoporosis. The treatment is most effective for increasing final adult height if it is initiated before age 6. Puberty resumes after discontinuation of the GnRH analogue. Counseling is an important aspect of the overall treatment strategy.
In children with gonadotropin-independent precocious puberty, inhibitors of steroidogenesis, such as ketoconazole, and AR antagonists have been used empirically. Long-term treatment with spironolactone (a weak androgen antagonist) and ketoconazole has been reported to normalize growth rate and bone maturation and to improve predicted height in small, nonrandomized trials in boys with familial male-limited precocious puberty. Aromatase inhibitors, such as testolactone and letrozole, have been used as an adjunct to antiandrogen and GnRH analogue therapy for children with familial male-limited precocious puberty, CAH, and McCune-Albright syndrome.
DELAYED PUBERTY
Puberty is delayed in boys if it has not ensued by age 14, an age that is 2–2.5 standard deviations above the mean for healthy children. Delayed puberty is more common in boys than in girls. There are four main categories of delayed puberty: (1) constitutional delay of growth and puberty (~60% of cases); (2) functional hypogonadotropic hypogonadism caused by systemic illness or malnutrition (~20% of cases); (3) hypogonadotropic hypogonadism caused by genetic or acquired defects in the hypothalamic-pituitary region (~10% of cases); and (4) hypergonadotropic hypogonadism secondary to primary gonadal failure (~15% of cases) (Table 411-1). Functional hypogonadotropic hypogonadism is more common in girls than in boys. Permanent causes of hypogonadotropic or hypergonadotropic hypogonadism are identified in >25% of boys with delayed puberty.
TREATMENT | DELAYED PUBERTY |
If therapy is considered appropriate, it can begin with 25–50 mg testosterone enanthate or testosterone cypionate every 2 weeks, or by using a 2.5-mg testosterone patch or 25-mg testosterone gel. Because aromatization of testosterone to estrogen is obligatory for mediating androgen effects on epiphyseal fusion, concomitant treatment with aromatase inhibitors may allow attainment of greater final adult height. Testosterone treatment should be interrupted after 6 months to determine if endogenous LH and FSH secretion have ensued. Other causes of delayed puberty should be considered when there are associated clinical features or when boys do not enter puberty spontaneously after a year of observation or treatment.
Reassurance without hormonal treatment is appropriate for many individuals with presumed constitutional delay of puberty. However, the impact of delayed growth and pubertal progression on a child’s social relationships and school performance should be weighed. Also, boys with constitutional delay of puberty are less likely to achieve their full genetic height potential and have reduced total-body bone mass as adults, mainly due to narrow limb bones and vertebrae as a result of impaired periosteal expansion during puberty. Administration of androgen therapy to boys with constitutional delay does not affect final height, and when administered with an aromatase inhibitor, it may improve final height.
DISORDERS OF THE MALE REPRODUCTIVE AXIS DURING ADULTHOOD
HYPOGONADOTROPIC HYPOGONADISM
Because LH and FSH are trophic hormones for the testes, impaired secretion of these pituitary gonadotropins results in secondary hypogonadism, which is characterized by low testosterone in the setting of low LH and FSH. Those with the most severe deficiency have complete absence of pubertal development, sexual infantilism, and, in some cases, hypospadias and undescended testes. Patients with partial gonadotropin deficiency have delayed or arrested sex development. The 24-h LH secretory profiles are heterogeneous in patients with hypogonadotropic hypogonadism, reflecting variable abnormalities of LH pulse frequency or amplitude. In severe cases, basal LH is low and there are no LH pulses. A smaller subset of patients has low-amplitude LH pulses or markedly reduced pulse frequency. Occasionally, only sleep-entrained LH pulses occur, reminiscent of the pattern seen in the early stages of puberty. Hypogonadotropic hypogonadism can be classified into congenital and acquired disorders. Congenital disorders most commonly involve GnRH deficiency, which leads to gonadotropin deficiency. Acquired disorders are much more common than congenital disorders and may result from a variety of sellar mass lesions or infiltrative diseases of the hypothalamus or pituitary.
Congenital Disorders Associated with Gonadotropin Deficiency Congenital hypogonadotropic hypogonadism is a heterogeneous group of disorders characterized by decreased gonadotropin secretion and testicular dysfunction either due to impaired function of the GnRH pulse generator or the gonadotrope. The disorders characterized by GnRH deficiency represent a family of oligogenic disorders whose phenotype spans a wide spectrum. Some individuals with GnRH deficiency may suffer from complete absence of pubertal development, while others may manifest varying degrees of gonadotropin deficiency and pubertal delay, and a subset that carries the same mutations as their affected family members may even have normal reproductive function. In approximately 10% of men with idiopathic hypogonadotropic hypogonadism, reversal of gonadotropin deficiency may occur in adult life after sex steroid therapy. Also, a small fraction of men with idiopathic hypogonadotropic hypogonadism may present with androgen deficiency and infertility in adult life after having gone through apparently normal pubertal development. Nutritional, emotional, or metabolic stress may unmask gonadotropin deficiency and reproductive dysfunction (analogous to hypothalamic amenorrhea) in some patients who harbor mutations in the candidate genes but who previously had normal reproductive function. The clinical phenotype may include isolated anosmia or hyposmia. These striking variations in phenotypic presentation of GnRH deficiency have highlighted the important role of oligogenicity and gene-gene and gene-environment interactions in shaping the clinical phenotype.
Mutations in a number of genes involved in the development and migration of GnRH neurons or in the regulation of GnRH secretion have been linked to GnRH deficiency, although the genetic defect remains elusive in nearly two-thirds of cases. Familial hypogonadotropic hypogonadism can be transmitted as an X-linked (20%), autosomal recessive (30%), or autosomal dominant (50%) trait. Some individuals with idiopathic hypogonadotropic hypogonadism (IHH) have sporadic mutations in the same genes that cause inherited forms of the disorder. The genetic defects associated with GnRH deficiency can be conveniently classified as anosmic (Kallmann’s syndrome) or normosmic (Table 411-2), although the occurrence of both anosmic and normosmic forms of GnRH deficiency in the same families suggests commonality of pathophysiologic mechanisms. Kallmann’s syndrome, the anosmic form of GnRH deficiency, can result from mutations in one or more genes associated with olfactory bulb morphogenesis and the migration of GnRH neurons from their origin in the region of the olfactory placode, along the scaffold established by the olfactory nerves, through the cribriform plate into their final location into the preoptic region of the hypothalamus. Thus, mutations in KAL1, FGF8, FGFR1, NELF, PROK2, PROK2R, and CHD7 have been described in patients with Kallmann’s syndrome. An X-linked form of IHH is caused by mutations in the KAL1 gene, which encodes anosmin, a protein that mediates the migration of neural progenitors of the olfactory bulb and GnRH-producing neurons. These individuals have GnRH deficiency and variable combinations of anosmia or hyposmia, renal defects, and neurologic abnormalities including mirror movements. Mutations in the FGFR1 gene cause an autosomal dominant form of hypogonadotropic hypogonadism that clinically resembles Kallmann’s syndrome; mutations in its putative ligand, FGF8 gene product, have also been associated with IHH. Prokineticin 2 (PROK2) also encodes a protein involved in migration and development of olfactory and GnRH neurons. Recessive mutations in PROK2 or in its receptor, PROKR2, have been associated with both anosmic and normosmic forms of hypogonadotropic hypogonadism.
CAUSES OF CONGENITAL HYPOGONADOTROPIC HYPOGONADISM |
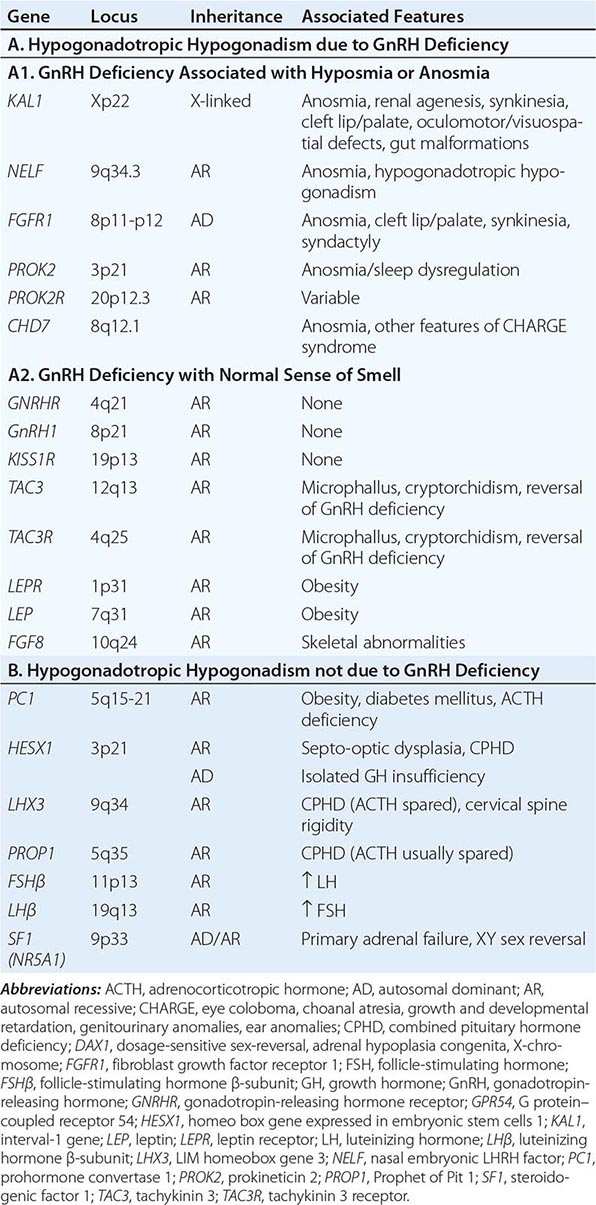
Normosmic GnRH deficiency results from defects in pulsatile GnRH secretion, its regulation, or its action on the gonadotrope and has been associated with mutations in GnRHR, GNRH1, KISS1R, TAC3, TACR3, and NROB1 (DAX1). Some mutations, such as those in PROK2, PROKR2, and CHD7, have been associated with both the anosmic and normosmic forms of IHH. GnRHR mutations, the most frequent identifiable cause of normosmic IHH, account for ~40% of autosomal recessive and 10% of sporadic cases of hypogonadotropic hypogonadism. These patients have decreased LH response to exogenous GnRH. Some receptor mutations alter GnRH binding affinity, allowing apparently normal responses to pharmacologic doses of exogenous GnRH, whereas other mutations may alter signal transduction downstream of hormone binding. Mutations of the GnRH1 gene have also been reported in patients with hypogonadotropic hypogonadism, although they are rare. G protein–coupled receptor KISS1R (GPR54) and its cognate ligand, kisspeptin (KISS1), are important regulators of sexual maturation in primates. Recessive mutations in GPR54 cause gonadotropin deficiency without anosmia. Patients retain responsiveness to exogenous GnRH, suggesting an abnormality in the neural pathways controlling GnRH release. The genes encoding neurokinin B (TAC3), which is involved in preferential activation of GnRH release in early development, and its receptor (TAC3R) have been implicated in some families with normosmic IHH. Mutations in more than one gene (digenicity or oligogenicity) may contribute to clinical heterogeneity in IHH patients. X-linked hypogonadotropic hypogonadism also occurs in adrenal hypoplasia congenita, a disorder caused by mutations in the DAX1 gene, which encodes a nuclear receptor in the adrenal gland and reproductive axis. Adrenal hypoplasia congenita is characterized by absent development of the adult zone of the adrenal cortex, leading to neonatal adrenal insufficiency. Puberty usually does not occur or is arrested, reflecting variable degrees of gonadotropin deficiency. Although sexual differentiation is normal, most patients have testicular dysgenesis and impaired spermatogenesis despite gonadotropin replacement. Less commonly, adrenal hypoplasia congenita, sex reversal, and hypogonadotropic hypogonadism can be caused by mutations of steroidogenic factor 1 (SF1). Rarely, recessive mutations in the LHβ or FSHβ gene have been described in patients with selective deficiencies of these gonadotropins. In approximately 10% of men with IHH, reversal of gonadotropin deficiency may occur in adult life. Also, a small fraction of men with IHH may present with androgen deficiency and infertility in adult life after having gone through apparently normal pubertal development.
A number of homeodomain transcription factors are involved in the development and differentiation of the specialized hormone-producing cells within the pituitary gland (Table 411-2). Patients with mutations of PROP1 have combined pituitary hormone deficiency that includes GH, prolactin (PRL), thyroid-stimulating hormone (TSH), LH, and FSH, but not ACTH. LHX3 mutations cause combined pituitary hormone deficiency in association with cervical spine rigidity. HESX1 mutations cause septo-optic dysplasia and combined pituitary hormone deficiency.
Prader-Willi syndrome is characterized by obesity, hypotonic musculature, mental retardation, hypogonadism, short stature, and small hands and feet. Prader-Willi syndrome is a genomic imprinting disorder caused by deletions of the proximal portion of the paternally derived chromosome 15q11-15q13 region, which contains a bipartite imprinting center, uniparental disomy of the maternal alleles, or mutations of the genes/loci involved in imprinting (Chap. 83e). Laurence-Moon syndrome is an autosomal recessive disorder characterized by obesity, hypogonadism, mental retardation, polydactyly, and retinitis pigmentosa. Recessive mutations of leptin, or its receptor, cause severe obesity and pubertal arrest, apparently because of hypothalamic GnRH deficiency (Chap. 415e).
Acquired Hypogonadotropic Disorders • SEVERE ILLNESS, STRESS, MALNUTRITION, AND EXERCISE These factors may cause reversible gonadotropin deficiency. Although gonadotropin deficiency and reproductive dysfunction are well documented in these conditions in women, men exhibit similar but less pronounced responses. Unlike women, most male runners and other endurance athletes have normal gonadotropin and sex steroid levels, despite low body fat and frequent intensive exercise. Testosterone levels fall at the onset of illness and recover during recuperation. The magnitude of gonadotropin suppression generally correlates with the severity of illness. Although hypogonadotropic hypogonadism is the most common cause of androgen deficiency in patients with acute illness, some have elevated levels of LH and FSH, which suggest primary gonadal dysfunction. The pathophysiology of reproductive dysfunction during acute illness is unknown but likely involves a combination of cytokine and/or glucocorticoid effects. There is a high frequency of low testosterone levels in patients with chronic illnesses such as HIV infection, end-stage renal disease, chronic obstructive lung disease, and many types of cancer and in patients receiving glucocorticoids. About 20% of HIV-infected men with low testosterone levels have elevated LH and FSH levels; these patients presumably have primary testicular dysfunction. The remaining 80% have either normal or low LH and FSH levels; these men have a central hypothalamic-pituitary defect or a dual defect involving both the testis and the hypothalamic-pituitary centers. Muscle wasting is common in chronic diseases associated with hypogonadism, which also leads to debility, poor quality of life, and adverse outcome of disease. There is great interest in exploring strategies that can reverse androgen deficiency or attenuate the sarcopenia associated with chronic illness.
Men using opioids for relief of cancer or noncancerous pain or because of addiction often have suppressed testosterone and LH levels and high prevalence of sexual dysfunction and osteoporosis; the degree of suppression is dose-related and particularly severe with long-acting opioids such as methadone. Opioids suppress GnRH secretion and alter the sensitivity to feedback inhibition by gonadal steroids. Men who are heavy users of marijuana have decreased testosterone secretion and sperm production. The mechanism of marijuana-induced hypogonadism is decreased GnRH secretion. Gynecomastia observed in marijuana users can also be caused by plant estrogens in crude preparations. Androgen deprivation therapy in men with prostate cancer has been associated with increased risk of bone fractures, diabetes mellitus, cardiovascular events, fatigue, sexual dysfunction, and poor quality of life.
OBESITY In men with mild to moderate obesity, SHBG levels decrease in proportion to the degree of obesity, resulting in lower total testosterone levels. However, free testosterone levels usually remain within the normal range. The decrease in SHBG levels is caused by increased circulating insulin, which inhibits SHBG production. Estradiol levels are higher in obese men compared to healthy, nonobese controls, because of aromatization of testosterone to estradiol in adipose tissue. Weight loss is associated with reversal of these abnormalities including an increase in total and free testosterone levels and a decrease in estradiol levels. A subset of obese men with moderate to severe obesity may have a defect in the hypothalamic-pituitary axis as suggested by low free testosterone in the absence of elevated gonadotropins. Weight gain in adult men can accelerate the rate of age-related decline in testosterone levels.
HYPERPROLACTINEMIA (See also Chap. 403) Elevated PRL levels are associated with hypogonadotropic hypogonadism. PRL inhibits hypothalamic GnRH secretion either directly or through modulation of tuberoinfundibular dopaminergic pathways. A PRL-secreting tumor may also destroy the surrounding gonadotropes by invasion or compression of the pituitary stalk. Treatment with dopamine agonists reverses gonadotropin deficiency, although there may be a delay relative to PRL suppression.
SELLAR MASS LESIONS Neoplastic and nonneoplastic lesions in the hypothalamus or pituitary can directly or indirectly affect gonadotrope function. In adults, pituitary adenomas constitute the largest category of space-occupying lesions affecting gonadotropin and other pituitary hormone production. Pituitary adenomas that extend into the suprasellar region can impair GnRH secretion and mildly increase PRL secretion (usually <50 μg/L) because of impaired tonic inhibition by dopaminergic pathways. These tumors should be distinguished from prolactinomas, which typically secrete higher PRL levels. The presence of diabetes insipidus suggests the possibility of a craniopharyngioma, infiltrative disorder, or other hypothalamic lesions (Chap. 404).
HEMOCHROMATOSIS (See also Chap. 428) Both the pituitary and testis can be affected by excessive iron deposition. However, the pituitary defect is the predominant lesion in most patients with hemochromatosis and hypogonadism. The diagnosis of hemochromatosis is suggested by the association of characteristic skin discoloration, hepatic enlargement or dysfunction, diabetes mellitus, arthritis, cardiac conduction defects, and hypogonadism.
PRIMARY TESTICULAR CAUSES OF HYPOGONADISM
Common causes of primary testicular dysfunction include Klinefelter’s syndrome, uncorrected cryptorchidism, cancer chemotherapy, radiation to the testes, trauma, torsion, infectious orchitis, HIV infection, anorchia syndrome, and myotonic dystrophy. Primary testicular disorders may be associated with impaired spermatogenesis, decreased androgen production, or both. See Chap. 410 for disorders of testis development, androgen synthesis, and androgen action.
Klinefelter’s Syndrome (See also Chap. 410) Klinefelter’s syndrome is the most common chromosomal disorder associated with testicular dysfunction and male infertility. It occurs in about 1 in 600 live-born males. Azoospermia is the rule in men with Klinefelter’s syndrome who have the 47,XXY karyotype; however, men with mosaicism may have germ cells, especially at a younger age. The clinical phenotype of Klinefelter’s syndrome can be heterogeneous possibly because of mosaicism, polymorphisms in AR gene, variable testosterone levels, or other genetic factors. Testicular histology shows hyalinization of seminiferous tubules and absence of spermatogenesis. Although their function is impaired, the number of Leydig cells appears to increase. Testosterone is decreased and estradiol is increased, leading to clinical features of undervirilization and gynecomastia. Men with Klinefelter’s syndrome are at increased risk of systemic lupus erythematosus, Sjögren’s syndrome, breast cancer, diabetes mellitus, osteoporosis, non-Hodgkin’s lymphoma, and lung cancer, and reduced risk of prostate cancer. Periodic mammography for breast cancer surveillance is recommended for men with Klinefelter’s syndrome. Fertility has been achieved by intracytoplasmic injection of sperm retrieved surgically from testicular biopsies of men with Klinefelter’s syndrome, including some men with the nonmosaic form of Klinefelter’s syndrome. The karyotypes 48,XXXY and 49,XXXXY are associated with a more severe phenotype, increased risk of congenital malformations, and lower intelligence than 47,XXY individuals.
Cryptorchidism Cryptorchidism occurs when there is incomplete descent of the testis from the abdominal cavity into the scrotum. About 3% of full-term and 30% of premature male infants have at least one undescended testis at birth, but descent is usually complete by the first few weeks of life. The incidence of cryptorchidism is <1% by 9 months of age. Androgens regulate predominantly the inguinoscrotal descent of the testes through degeneration of the craniosuspensory ligament and a shortening of the gubernaculums, respectively. Mutations in INSL3 and leucine-rich repeat family of G protein–coupled receptor 8 (LGR8), which regulate the transabdominal portion of testicular descent, have been found in some patients with cryptorchidism.
Cryptorchidism is associated with increased risk of malignancy, infertility, inguinal hernia, and torsion. Unilateral cryptorchidism, even when corrected before puberty, is associated with decreased sperm count, possibly reflecting unrecognized damage to the fully descended testis or other genetic factors. Epidemiologic, clinical, and molecular evidence supports the idea that cryptorchidism, hypospadias, impaired spermatogenesis, and testicular cancer may be causally related to common genetic and environment perturbations and are components of the testicular dysgenesis syndrome.
Acquired Testicular Defects Viral orchitis may be caused by the mumps virus, echovirus, lymphocytic choriomeningitis virus, and group B arboviruses. Orchitis occurs in as many as one-fourth of adult men with mumps; the orchitis is unilateral in about two-thirds and bilateral in the remainder. Orchitis usually develops a few days after the onset of parotitis but may precede it. The testis may return to normal size and function or undergo atrophy. Semen analysis returns to normal for three-fourths of men with unilateral involvement but for only one-third of men with bilateral orchitis. Trauma, including testicular torsion, can also cause secondary atrophy of the testes. The exposed position of the testes in the scrotum renders them susceptible to both thermal and physical trauma, particularly in men with hazardous occupations.
The testes are sensitive to radiation damage. Doses >200 mGy (20 rad) are associated with increased FSH and LH levels and damage to the spermatogonia. After ~800 mGy (80 rad), oligospermia or azoospermia develops, and higher doses may obliterate the germinal epithelium. Permanent androgen deficiency in adult men is uncommon after therapeutic radiation; however, most boys given direct testicular radiation therapy for acute lymphoblastic leukemia have permanently low testosterone levels. Sperm banking should be considered before patients undergo radiation treatment or chemotherapy.
Drugs interfere with testicular function by several mechanisms, including inhibition of testosterone synthesis (e.g., ketoconazole), blockade of androgen action (e.g., spironolactone), increased estrogen (e.g., marijuana), or direct inhibition of spermatogenesis (e.g., chemotherapy).
Combination chemotherapy for acute leukemia, Hodgkin’s disease, and testicular and other cancers may impair Leydig cell function and cause infertility. The degree of gonadal dysfunction depends on the type of chemotherapeutic agent and the dose and duration of therapy. Because of high response rates and the young age of these men, infertility and androgen deficiency have emerged as important long-term complications of cancer chemotherapy. Cyclophosphamide and combination regimens containing procarbazine are particularly toxic to germ cells. Thus, 90% of men with Hodgkin’s lymphoma receiving MOPP (mechlorethamine, vincristine, procarbazine, prednisone) therapy develop azoospermia or extreme oligozoospermia; newer regimens that do not include procarbazine, such as ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine), are less toxic to germ cells.
Alcohol, when consumed in excess for prolonged periods, decreases testosterone, independent of liver disease or malnutrition. Elevated estradiol and decreased testosterone levels may occur in men taking digitalis.
The occupational and recreational history should be carefully evaluated in all men with infertility because of the toxic effects of many chemical agents on spermatogenesis. Known environmental hazards include pesticides (e.g., vinclozolin, dicofol, atrazine), sewage contaminants (e.g., ethinyl estradiol in birth control pills, surfactants such as octylphenol, nonyphenol), plasticizers (e.g., pthalates), flame retardants (e.g., polychlorinated biphenyls, polybrominated diphenol ethers), industrial pollutants (e.g., heavy metals cadmium and lead, dioxins, polycyclic aromatic hydrocarbons), microwaves, and ultrasound. In some populations, sperm density is said to have declined by as much as 40% in the past 50 years. Environmental estrogens or antiandrogens may be partly responsible.
Testicular failure also occurs as a part of polyglandular autoimmune insufficiency (Chap. 408). Sperm antibodies can cause isolated male infertility. In some instances, these antibodies are secondary phenomena resulting from duct obstruction or vasectomy. Granulomatous diseases can affect the testes, and testicular atrophy occurs in 10–20% of men with lepromatous leprosy because of direct tissue invasion by the mycobacteria. The tubules are involved initially, followed by endarteritis and destruction of Leydig cells.
Systemic disease can cause primary testis dysfunction in addition to suppressing gonadotropin production. In cirrhosis, a combined testicular and pituitary abnormality leads to decreased testosterone production independent of the direct toxic effects of ethanol. Impaired hepatic extraction of adrenal androstenedione leads to extraglandular conversion to estrone and estradiol, which partially suppresses LH. Testicular atrophy and gynecomastia are present in approximately one-half of men with cirrhosis. In chronic renal failure, androgen synthesis and sperm production decrease despite elevated gonadotropins. The elevated LH level is due to reduced clearance, but it does not restore normal testosterone production. About one-fourth of men with renal failure have hyperprolactinemia. Improvement in testosterone production with hemodialysis is incomplete, but successful renal transplantation may return testicular function to normal. Testicular atrophy is present in one-third of men with sickle cell anemia. The defect may be at either the testicular or the hypothalamic-pituitary level. Sperm density can decrease temporarily after acute febrile illness in the absence of a change in testosterone production. Infertility in men with celiac disease is associated with a hormonal pattern typical of androgen resistance, namely elevated testosterone and LH levels.
Neurologic diseases associated with altered testicular function include myotonic dystrophy, spinobulbar muscular atrophy, and paraplegia. In myotonic dystrophy, small testes may be associated with impairment of both spermatogenesis and Leydig cell function. Spinobulbar muscular atrophy is caused by an expansion of the glutamine repeat sequences in the amino-terminal region of the AR; this expansion impairs function of the AR, but it is unclear how the alteration is related to the neurologic manifestations. Men with spinobulbar muscular atrophy often have undervirilization and infertility as a late manifestation. Spinal cord lesions that cause paraplegia can lead to a temporary decrease in testosterone levels and may cause persistent defects in spermatogenesis; some patients retain the capacity for penile erection and ejaculation.
ANDROGEN INSENSITIVITY SYNDROMES
Mutations in the AR cause resistance to the action of testosterone and DHT. These X-linked mutations are associated with variable degrees of defective male phenotypic development and undervirilization (Chap. 410). Although not technically hormone-insensitivity syndromes, two genetic disorders impair testosterone conversion to active sex steroids. Mutations in the SRD5A2 gene, which encodes 5α-reductase type 2, prevent the conversion of testosterone to DHT, which is necessary for the normal development of the male external genitalia. Mutations in the CYP19 gene, which encodes aromatase, prevent testosterone conversion to estradiol. Males with CYP19 mutations have delayed epiphyseal fusion, tall stature, eunuchoid proportions, and osteoporosis, consistent with evidence from an estrogen receptor–deficient individual that these testosterone actions are mediated indirectly via estrogen.
GYNECOMASTIA
Gynecomastia refers to enlargement of the male breast. It is caused by excess estrogen action and is usually the result of an increased estrogen-to-androgen ratio. True gynecomastia is associated with glandular breast tissue that is >4 cm in diameter and often tender. Glandular tissue enlargement should be distinguished from excess adipose tissue: glandular tissue is firmer and contains fibrous-like cords. Gynecomastia occurs as a normal physiologic phenomenon in the newborn (due to transplacental transfer of maternal and placental estrogens), during puberty (high estrogen-to-androgen ratio in early stages of puberty), and with aging (increased fat tissue and increased aromatase activity), but it can also result from pathologic conditions associated with androgen deficiency or estrogen excess. The prevalence of gynecomastia increases with age and body mass index (BMI), likely because of increased aromatase activity in adipose tissue. Medications that alter androgen metabolism or action may also cause gynecomastia. The relative risk of breast cancer is increased in men with gynecomastia, although the absolute risk is relatively small.
PATHOLOGIC GYNECOMASTIA
Any cause of androgen deficiency can lead to gynecomastia, reflecting an increased estrogen-to-androgen ratio, because estrogen synthesis still occurs by aromatization of residual adrenal and gonadal androgens. Gynecomastia is a characteristic feature of Klinefelter’s syndrome (Chap. 410). Androgen insensitivity disorders also cause gynecomastia. Excess estrogen production may be caused by tumors, including Sertoli cell tumors in isolation or in association with Peutz-Jeghers syndrome or Carney complex. Tumors that produce hCG, including some testicular tumors, stimulate Leydig cell estrogen synthesis. Increased conversion of androgens to estrogens can be a result of increased availability of substrate (androstenedione) for extraglandular estrogen formation (CAH, hyperthyroidism, and most feminizing adrenal tumors) or of diminished catabolism of androstenedione (liver disease) so that estrogen precursors are shunted to aromatase in peripheral sites. Obesity is associated with increased aromatization of androgen precursors to estrogens. Extraglandular aromatase activity can also be increased in tumors of the liver or adrenal gland or rarely as an inherited disorder. Several families with increased peripheral aromatase activity inherited as an autosomal dominant or as an X-linked disorder have been described. In some families with this disorder, an inversion in chromosome 15q21.2-3 causes the CYP19 gene to be activated by the regulatory elements of contiguous genes, resulting in excessive estrogen production in the fat and other extragonadal tissues. Drugs can cause gynecomastia by acting directly as estrogenic substances (e.g., oral contraceptives, phytoestrogens, digitalis) or by inhibiting androgen synthesis (e.g., ketoconazole) or action (e.g., spironolactone).
Because up to two-thirds of pubertal boys and half of hospitalized men have palpable glandular tissue that is benign, detailed investigation or intervention is not indicated in all men presenting with gynecomastia (Fig. 411-5). In addition to the extent of gynecomastia, recent onset, rapid growth, tender tissue, and occurrence in a lean subject should prompt more extensive evaluation. This should include a careful drug history, measurement and examination of the testes, assessment of virilization, evaluation of liver function, and hormonal measurements including testosterone, estradiol, and androstenedione, LH, and hCG. A karyotype should be obtained in men with very small testes to exclude Klinefelter’s syndrome. Despite extensive evaluation, the etiology is established in fewer than one-half of patients.
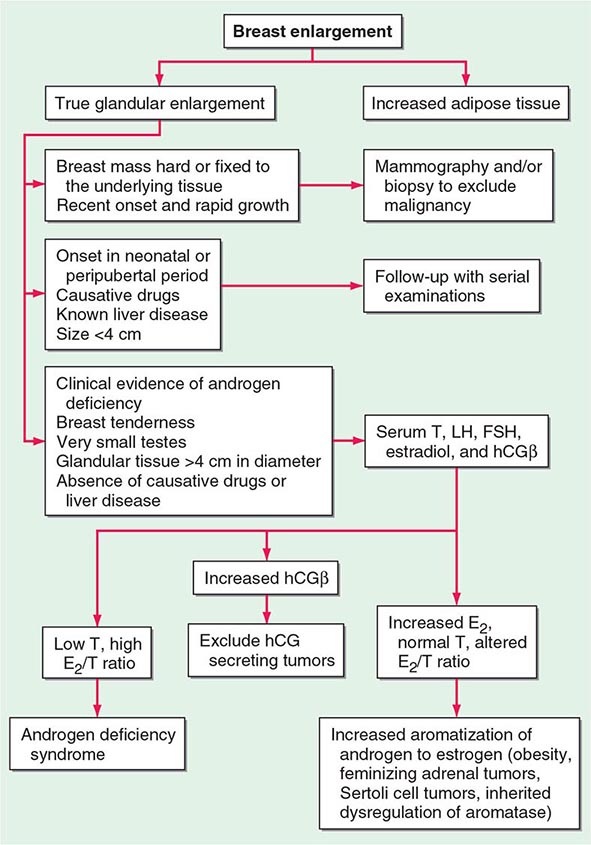
FIGURE 411-5 Evaluation of gynecomastia. E2, 17β-estradiol; hCGβ, human chorionic gonadotropin β; T, testosterone.
TREATMENT | GYNECOMASTIA |
When the primary cause can be identified and corrected, breast enlargement usually subsides over several months. However, if gynecomastia is of long duration, surgery is the most effective therapy. Indications for surgery include severe psychological and/or cosmetic problems, continued growth or tenderness, or suspected malignancy. In patients who have painful gynecomastia and in whom surgery cannot be performed, treatment with antiestrogens such as tamoxifen (20 mg/d) can reduce pain and breast tissue size in over half the patients. Estrogen receptor antagonists, tamoxifen and raloxifene, have been reported in small trials to reduce breast size in men with pubertal gynecomastia, although complete regression of breast enlargement is unusual with the use of estrogen receptor antagonists. Aromatase inhibitors can be effective in the early proliferative phase of the disorder. However, in a randomized trial in men with established gynecomastia, anastrozole proved no more effective than placebo in reducing breast size. Tamoxifen is effective in the prevention and treatment of breast enlargement and breast pain in men with prostate cancer who are receiving antiandrogen therapy.
AGING-RELATED CHANGES IN MALE REPRODUCTIVE FUNCTION
A number of cross-sectional and longitudinal studies (e.g., The Baltimore Longitudinal Study of Aging, the Framingham Heart Study, the Massachusetts Male Aging Study, and the European Male Aging Study) have established that testosterone concentrations decrease with advancing age. This age-related decline starts in the third decade of life and progresses slowly; the rate of decline in testosterone concentrations is greater in obese men, men with chronic illness, and those taking medications than in healthy older men. Because SHBG concentrations are higher in older men than in younger men, free or bioavailable testosterone concentrations decline with aging to a greater extent than total testosterone concentrations. The age-related decline in testosterone is due to defects at all levels of the hypothalamic-pituitary-testicular axis: pulsatile GnRH secretion is attenuated, LH response to GnRH is reduced, and testicular response to LH is impaired. However, the gradual rise of LH with aging suggests that testis dysfunction is the main cause of declining androgen levels. The term andropause has been used to denote age-related decline in testosterone concentrations; this term is a misnomer because there is no discrete time when testosterone concentrations decline abruptly. The approach to evaluating hypogonadism is summarized in Fig. 411-6.
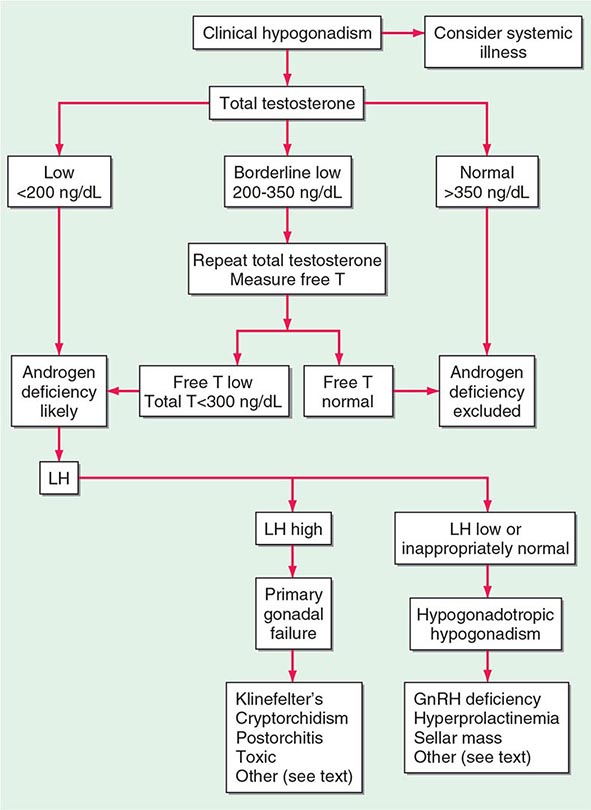
FIGURE 411-6 Evaluation of hypogonadism. GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone; T, testosterone.
In epidemiologic surveys, low total and bioavailable testosterone concentrations have been associated with decreased appendicular skeletal muscle mass and strength, decreased self-reported physical function, higher visceral fat mass, insulin resistance, and increased risk of coronary artery disease and mortality, although the associations are weak. An analysis of signs and symptoms in older men in the European Male Aging Study revealed a syndromic association of sexual symptoms with total testosterone levels below 320 ng/dL and free testosterone levels below 64 pg/mL in community-dwelling older men. In systematic reviews of randomized controlled trials, testosterone therapy of healthy older men with low or low-normal testosterone levels was associated with greater increments in lean body mass, grip strength, and self-reported physical function compared with placebo. Testosterone therapy also induced greater improvement in vertebral but not femoral bone mineral density. Testosterone therapy of older men with sexual dysfunction and unequivocally low testosterone levels improves libido, but testosterone effects on erectile function and response to selective phosphodiesterase inhibitors have been inconsistent. Testosterone therapy has not been shown to improve depression scores, fracture risk, cognitive function, response to phosphodiesterase inhibitors, or clinical outcomes in older men. Furthermore, neither the long-term risks nor clinical benefits of testosterone therapy in older men have been demonstrated in adequately powered trials. Although there is no evidence that testosterone causes prostate cancer, there is concern that testosterone therapy might cause subclinical prostate cancers to grow. Testosterone therapy is associated with increased risk of detection of prostate events (Fig. 411-7).
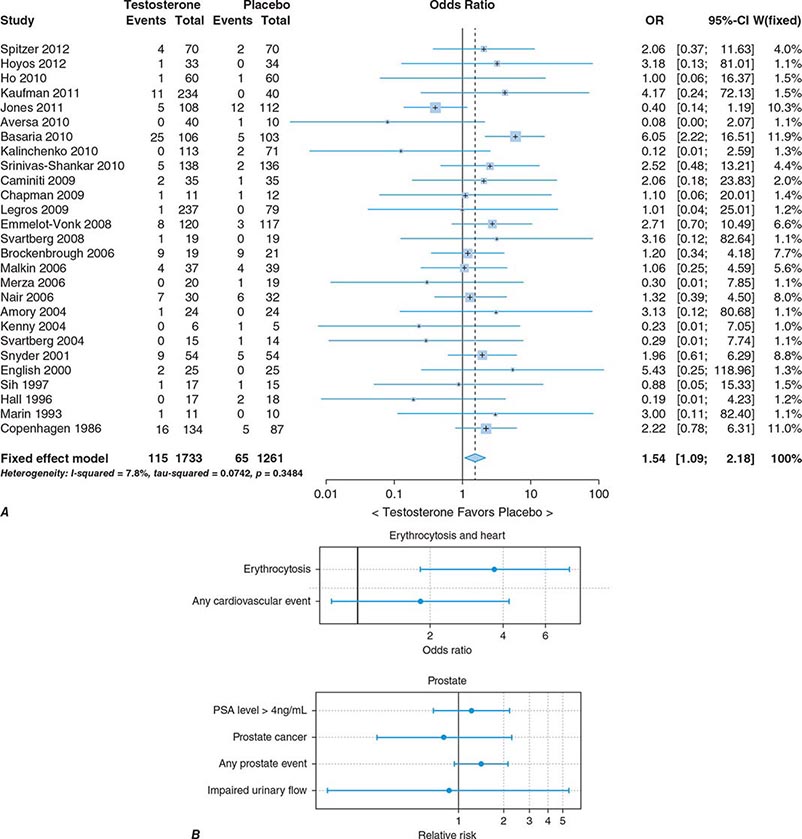
FIGURE 411-7 Meta-analyses of cardiovascular and prostate adverse events associated with testosterone therapy. A. A meta-analysis of cardiovascular-related events in randomized testosterone trials of 12 weeks or longer in duration. Randomization to testosterone was associated with a significantly increased risk of cardiovascular-related event (odds ratio [OR] 1.54). (Modified with permission from L Xu et al: Testosterone therapy and cardiovascular events among men: a systematic review and meta-analysis of placebo-controlled randomized trials BMC Med 11:108, 2013.) B. The relative risk of prostate events and the associated 95% confidence intervals (CIs) in a meta-analysis of randomized testosterone trials. PSA, prostate-specific antigen. (Data were derived from a meta-analysis by MM Fernández-Balsells et al: J Clin Endocrinol Metab 95:2560, 2010, and the figure was reproduced with permission from M Spitzer et al: Nat Rev Endocrinol 9:414, 2013.)
One randomized testosterone trial in older men with mobility limitation and high burden of chronic conditions, such as diabetes, heart disease, hypertension, and hyperlipidemia, reported a greater number of cardiovascular events in men randomized to the testosterone arm of the study than in those randomized to the placebo arm. Since then, two large retrospective analyses of patient databases have reported higher frequency of cardiovascular events, including myocardial infarction, in older men with preexisting heart disease (Fig. 411-7).
Population screening of all older men for low testosterone levels is not recommended, and testing should be restricted to men who have symptoms or physical features attributable to androgen deficiency. Testosterone therapy is not recommended for all older men with low testosterone levels. In older men with significant symptoms of androgen deficiency who have testosterone levels below 200 ng/dL, testosterone therapy may be considered on an individualized basis and should be instituted after careful discussion of the risks and benefits (see “Testosterone Replacement,” below).
Testicular morphology, semen production, and fertility are maintained up to a very old age in men. Although concern has been expressed about age-related increases in germ cell mutations and impairment of DNA repair mechanisms, there is no clear evidence that the frequency of chromosomal aneuploidy is increased in the sperm of older men. However, the incidence of autosomal dominant diseases, such as achondroplasia, polyposis coli, Marfan’s syndrome, and Apert’s syndrome, increases in the offspring of men who are advanced in age, consistent with transmission of sporadic missense mutations. Advanced paternal age may be associated with increased rates of de novo mutations, which may contribute to an increased risk of neurodevelopmental diseases such as schizophrenia and autism. The somatic mutations in male germ cells that enhance the proliferation of germ cells could lead to within-testis expansion of mutant clonal lines, thus favoring the propagation of germ cells carrying these pathogenic mutations and increasing the risk of mutations in the offspring of older fathers (the “selfish spermatogonial selection” hypothesis).
TREATMENT | ANDROGEN DEFICIENCY |
GONADOTROPINS
Gonadotropin therapy is used to establish or restore fertility in patients with gonadotropin deficiency of any cause. Several gonadotropin preparations are available. Human menopausal gonadotropin (hMG; purified from the urine of postmenopausal women) contains 75 IU FSH and 75 IU LH per vial. hCG (purified from the urine of pregnant women) has little FSH activity and resembles LH in its ability to stimulate testosterone production by Leydig cells. Recombinant LH is now available. Because of the expense of hMG, treatment is usually begun with hCG alone, and hMG is added later to promote the FSH-dependent stages of spermatid development. Recombinant human FSH (hFSH) is now available and is indistinguishable from purified urinary hFSH in its biologic activity and pharmacokinetics in vitro and in vivo, although the mature β subunit of recombinant hFSH has seven fewer amino acids. Recombinant hFSH is available in ampoules containing 75 IU (~7.5 μg FSH), which accounts for >99% of protein content. Once spermatogenesis is restored using combined FSH and LH therapy, hCG alone is often sufficient to maintain spermatogenesis.
Although a variety of treatment regimens are used, 1000–2000 IU of hCG or recombinant human LH (rhLH) administered intramuscularly three times weekly is a reasonable starting dose. Testosterone levels should be measured 6–8 weeks later and 48–72 h after the hCG or rhLH injection; the hCG/rhLH dose should be adjusted to achieve testosterone levels in the mid-normal range. Sperm counts should be monitored on a monthly basis. It may take several months for spermatogenesis to be restored; therefore, it is important to forewarn patients about the potential length and expense of the treatment and to provide conservative estimates of success rates. If testosterone levels are in the mid-normal range but the sperm concentrations are low after 6 months of therapy with hCG alone, FSH should be added. This can be done by using hMG, highly purified urinary hFSH, or recombinant hFSH. The selection of FSH dose is empirical. A common practice is to start with the addition of 75 IU FSH three times each week in conjunction with the hCG/rhLH injections. If sperm densities are still low after 3 months of combined treatment, the FSH dose should be increased to 150 IU. Occasionally, it may take ≥18–24 months for spermatogenesis to be restored.
The two best predictors of success using gonadotropin therapy in hypogonadotropic men are testicular volume at presentation and time of onset. In general, men with testicular volumes >8 mL have better response rates than those who have testicular volumes >4 mL. Patients who became hypogonadotropic after puberty experience higher success rates than those who have never undergone pubertal changes. Spermatogenesis can usually be reinitiated by hCG alone, with high rates of success for men with postpubertal onset of hypogonadotropism. The presence of a primary testicular abnormality, such as cryptorchidism, will attenuate testicular response to gonadotropin therapy. Prior androgen therapy does not preclude subsequent response to gonadotropin therapy, although some studies suggest that it may attenuate response to subsequent gonadotropin therapy.
TESTOSTERONE REPLACEMENT
Androgen therapy is indicated to restore testosterone levels to normal to correct features of androgen deficiency. Testosterone replacement improves libido and overall sexual activity; increases energy, lean muscle mass, and bone density; and decreases fat mass. The benefits of testosterone replacement therapy have only been proven in men who have documented androgen deficiency, as demonstrated by testosterone levels that are well below the lower limit of normal.
Testosterone is available in a variety of formulations with distinct pharmacokinetics (Table 411-3). Testosterone serves as a prohormone and is converted to 17β-estradiol by aromatase and to 5α-dihydrotestosterone by steroid 5α-reductase. Therefore, when evaluating testosterone formulations, it is important to consider whether the formulation being used can achieve physiologic estradiol and DHT concentrations, in addition to normal testosterone concentrations. Although testosterone concentrations at the lower end of the normal male range can restore sexual function, it is not clear whether low-normal testosterone levels can maintain bone mineral density and muscle mass. The current recommendation is to restore testosterone levels to the mid-normal range.
CLINICAL PHARMACOLOGY OF SOME TESTOSTERONE FORMULATIONS |
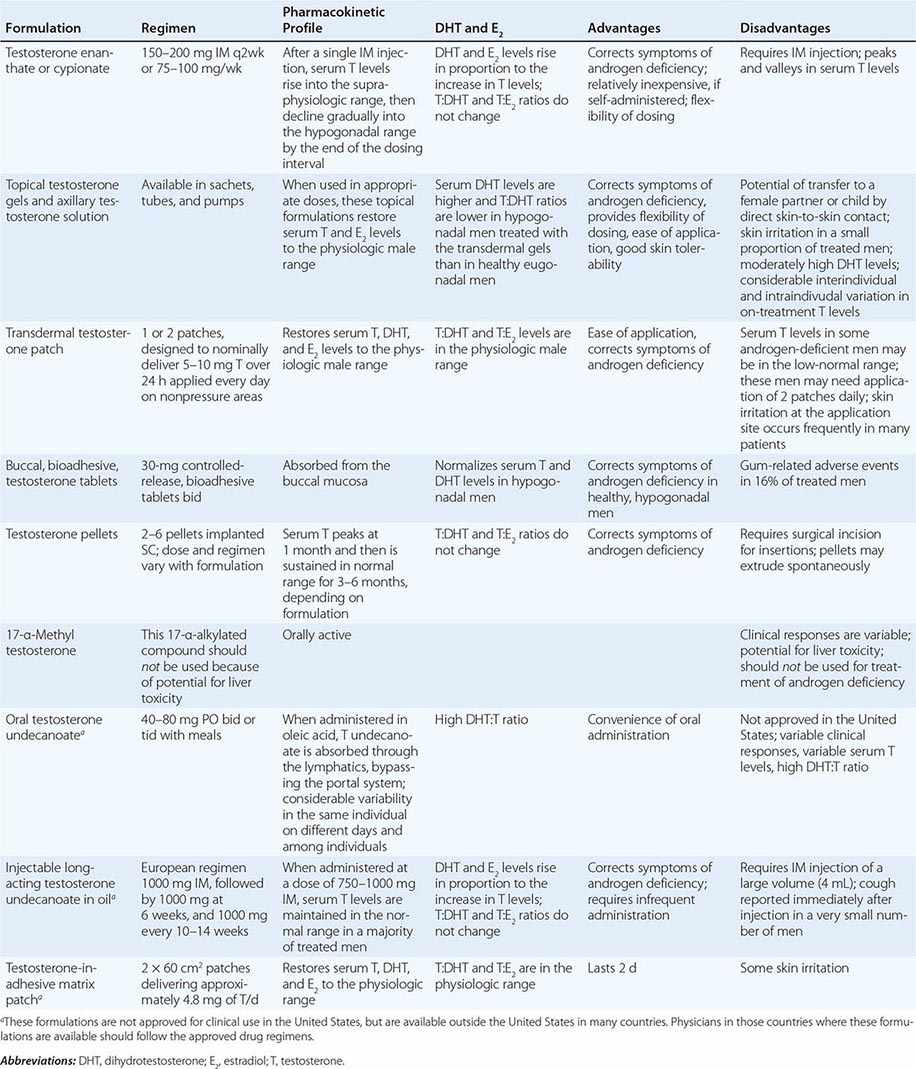
Oral Derivatives of Testosterone Testosterone is well-absorbed after oral administration but is quickly degraded during the first pass through the liver. Therefore, it is difficult to achieve sustained blood levels of testosterone after oral administration of crystalline testosterone. 17α-Alkylated derivatives of testosterone (e.g., 17α-methyl testosterone, oxandrolone, fluoxymesterone) are relatively resistant to hepatic degradation and can be administered orally; however, because of the potential for hepatotoxicity, including cholestatic jaundice, peliosis, and hepatoma, these formulations should not be used for testosterone replacement. Hereditary angioedema due to C1 esterase deficiency is the only exception to this general recommendation; in this condition, oral 17α-alkylated androgens are useful because they stimulate hepatic synthesis of the C1 esterase inhibitor.
Injectable Forms of Testosterone The esterification of testosterone at the 17β-hydroxy position makes the molecule hydrophobic and extends its duration of action. The slow release of testosterone ester from an oily depot in the muscle accounts for its extended duration of action. The longer the side chain, the greater is the hydrophobicity of the ester and the longer is the duration of action. Thus, testosterone enanthate, cypionate, and undecanoate with longer side chains have longer duration of action than testosterone propionate. Within 24 h after intramuscular administration of 200 mg testosterone enanthate or cypionate, testosterone levels rise into the high-normal or supraphysiologic range and then gradually decline into the hypogonadal range over the next 2 weeks. A bimonthly regimen of testosterone enanthate or cypionate therefore results in peaks and troughs in testosterone levels that are accompanied by changes in a patient’s mood, sexual desire, and energy level. The kinetics of testosterone enanthate and cypionate are similar. Estradiol and DHT levels are normal if testosterone replacement is physiologic.
Transdermal Testosterone Patch The nongenital testosterone patch, when applied in an appropriate dose, can normalize testosterone, DHT, and estradiol levels 4–12 h after application. Sexual function and well-being are restored in androgen-deficient men treated with the nongenital patch. One 5-mg patch may not be sufficient to increase testosterone into the mid-normal male range in all hypogonadal men; some patients may need two 5-mg patches daily to achieve the targeted testosterone concentrations. The use of testosterone patches may be associated with skin irritation in some individuals.
Testosterone Gel Several transdermal testosterone gels (e.g., Androgel, Testim, Fortesta, and Axiron), when applied topically to the skin in appropriate doses (Table 411-3), can maintain total and free testosterone concentrations in the normal range in hypogonadal men. The current recommendations are to begin with an initial U.S. Food and Drug Administration–approved dose and adjust the dose based on testosterone levels. The advantages of the testosterone gel include the ease of application and its flexibility of dosing. A major concern is the potential for inadvertent transfer of the gel to a sexual partner or to children who may come in close contact with the patient. The ratio of DHT to testosterone concentrations is higher in men treated with the testosterone gel than in healthy men. Also, there is considerable intra- and interindividual variation in serum testosterone levels in men treated with the transdermal gel due to variations in transdermal absorption and plasma clearance of testosterone. Therefore, monitoring of serum testosterone levels and multiple dose adjustments may be required to achieve and maintain testosterone levels in the target range.
Buccal Adhesive Testosterone A buccal testosterone tablet, which adheres to the buccal mucosa and releases testosterone as it is slowly dissolved, has been approved. After twice-daily application of 30-mg tablets, serum testosterone levels are maintained within the normal male range in a majority of treated hypogonadal men. The adverse effects include buccal ulceration and gum problems in a few subjects. The effects of food and brushing on absorption have not been studied in detail.
Implants of crystalline testosterone can be inserted in the subcutaneous tissue by means of a trocar through a small skin incision. Testosterone is released by surface erosion of the implant and absorbed into the systemic circulation. Two to six 200-mg implants can maintain testosterone in the mid- to high-normal range for up to 6 months. Potential drawbacks include incising the skin for insertion and removal and spontaneous extrusions and fibrosis at the site of the implant.
Testosterone Formulations Not Available in the United States Testosterone undecanoate, when administered orally in oleic acid, is absorbed preferentially through the lymphatics into the systemic circulation and is spared the first-pass degradation in the liver. Doses of 40–80 mg orally, two or three times daily, are typically used. However, the clinical responses are variable and suboptimal. DHT-to-testosterone ratios are higher in hypogonadal men treated with oral testosterone undecanoate, as compared to eugonadal men.
After initial priming, long-acting testosterone undecanoate in oil, when administered intramuscularly every 12 weeks, maintains serum testosterone, estradiol, and DHT in the normal male range and corrects symptoms of androgen deficiency in a majority of treated men. However, large injection volume (4 mL) is its relative drawback.
Novel Androgen Formulations A number of androgen formulations with better pharmacokinetics or more selective activity profiles are under development. A long-acting ester, testosterone undecanoate, when injected intramuscularly, can maintain circulating testosterone concentrations in the male range for 7–12 weeks. Initial clinical trials have demonstrated the feasibility of administering testosterone by the sublingual or buccal routes. 7α-Methyl-19-nortestosterone is an androgen that cannot be 5α-reduced; therefore, compared to testosterone, it has relatively greater agonist activity in muscle and gonadotropin suppression but lesser activity on the prostate.
Selective AR modulators (SARMs) are a class of AR ligands that bind the AR and display tissue-selective actions. A number of nonsteroidal SARMs that act as full agonists on the muscle and bone and that spare the prostate to varying degrees have advanced to phase 3 human trials. Nonsteroidal SARMs do not serve as substrates for either the steroid 5α-reductase or the CYP19 aromatase. SARM binding to AR induces specific conformational changes in the AR protein, which then modulates protein-protein interactions between AR and its coregulators, resulting in tissue-specific regulation of gene expression.
Pharmacologic Uses of Androgens Androgens and SARMs are being evaluated as anabolic therapies for functional limitations associated with aging and chronic illness. Testosterone supplementation increases skeletal muscle mass, maximal voluntary strength, and muscle power in healthy men, hypogonadal men, older men with low testosterone levels, HIV-infected men with weight loss, and men receiving glucocorticoids. These anabolic effects of testosterone are related to testosterone dose and circulating concentrations. Systematic reviews have confirmed that testosterone therapy of HIV-infected men with weight loss promotes improvements in body weight, lean body mass, muscle strength, and depression indices, leading to the recommendation that testosterone be considered as an adjunctive therapy in HIV-infected men who are experiencing unexplained weight loss and who have low testosterone levels. Similarly, in glucocorticoid-treated men, testosterone therapy should be considered to maintain muscle mass and strength and vertebral bone mineral density. It is unknown whether testosterone therapy of older men with functional limitations is safe and effective in improving physical function, vitality, and health-related quality of life and reducing disability. Concerns about potential adverse effects of testosterone on prostate and cardiovascular event rates have encouraged the development of SARMs that are preferentially anabolic and spare the prostate.
Testosterone administration induces hypertrophy of both type 1 and 2 fibers and increases satellite cell (muscle progenitor cells) and myonuclear number. Androgens promote the differentiation of mesenchymal, multipotent progenitor cells into the myogenic lineage and inhibit their differentiation into the adipogenic lineage. Testosterone may have additional effects on satellite cell replication and muscle protein synthesis, which may contribute to an increase in skeletal muscle mass.
Other indications for androgen therapy are in selected patients with anemia due to bone marrow failure (an indication largely supplanted by erythropoietin) or for hereditary angioedema.
Male Hormonal Contraception Based on Combined Administration of Testosterone and Gonadotropin Inhibitors Supraphysiologic doses of testosterone (200 mg testosterone enanthate weekly) suppress LH and FSH secretion and induce azoospermia in 50% of Caucasian men and >95% of Chinese men. The WHO-supported multicenter efficacy trials have demonstrated that suppression of spermatogenesis to azoospermia or severe oligozoospermia (<3 million/mL) by administration of testosterone enanthate to men results in highly effective contraception. Because of concern about long-term adverse effects of supraphysiologic testosterone doses, regimens that combine other gonadotropin inhibitors, such as GnRH antagonists and progestins with replacement doses of testosterone, are being investigated. Oral etonogestrel daily in combination with intramuscular testosterone decanoate every 4–6 weeks induced azoospermia or severe oligozoospermia (sperm density <1 million/mL) in 99% of treated men over a 1-year period. This regimen was associated with weight gain, deceased testicular volume, and decreased plasma high-density lipoprotein (HDL) cholesterol, and its long-term safety has not been demonstrated. SARMs that are more potent inhibitors of gonadotropins than testosterone and spare the prostate hold promise for their contraceptive potential.
Recommended Regimens for Androgen Replacement Testosterone esters are administered typically at doses of 75–100 mg intramuscularly every week, or 150–200 mg every 2 weeks. One or two 5-mg nongenital testosterone patches can be applied daily over the skin of the back, thigh, or upper arm away from pressure areas. Testosterone gels are typically applied over a covered area of skin at initial doses that vary with the formulation; patients should wash their hands after gel application. Bioadhesive buccal testosterone tablets at a dose of 30 mg are typically applied twice daily on the buccal mucosa.
Establishing Efficacy of Testosterone Replacement Therapy Because a clinically useful marker of androgen action is not available, restoration of testosterone levels to the mid-normal range remains the goal of therapy. Measurements of LH and FSH are not useful in assessing the adequacy of testosterone replacement. Testosterone should be measured 3 months after initiating therapy to assess adequacy of therapy. There is substantial interindividual variability in serum testosterone levels, especially with transdermal gels, presumably due to genetic differences in testosterone clearance and transdermal absorption. In patients who are treated with testosterone enanthate or cypionate, testosterone levels should be 350–600 ng/dL 1 week after the injection. If testosterone levels are outside this range, adjustments should be made either in the dose or in the interval between injections. In men on transdermal patch, gel, or buccal testosterone therapy, testosterone levels should be in the mid-normal range (500–700 ng/dL) 4–12 h after application. If testosterone levels are outside this range, the dose should be adjusted. Multiple dose adjustments are often necessary to achieve testosterone levels in the desired therapeutic range.
Restoration of sexual function, secondary sex characteristics, energy, and well-being and maintenance of muscle and bone health are important objectives of testosterone replacement therapy. The patient should be asked about sexual desire and activity, the presence of early morning erections, and the ability to achieve and maintain erections adequate for sexual intercourse. Some hypogonadal men continue to complain about sexual dysfunction even after testosterone replacement has been instituted; these patients may benefit from counseling. The hair growth in response to androgen replacement is variable and depends on ethnicity. Hypogonadal men with prepubertal onset of androgen deficiency who begin testosterone therapy in their late twenties or thirties may find it difficult to adjust to their newly found sexuality and may benefit from counseling. If the patient has a sexual partner, the partner should be included in counseling because of the dramatic physical and sexual changes that occur with androgen treatment.
Contraindications for Androgen Administration Testosterone administration is contraindicated in men with a history of prostate or breast cancer (Table 411-4). Testosterone therapy should not be administered without further urologic evaluation to men with a palpable prostate nodule or induration; to men with prostate-specific antigen levels >4 ng/mL or >3 ng/mL in men at high risk for prostate cancer such as African Americans or men with first-degree relatives with prostate cancer; or to men with severe lower urinary tract symptoms (American Urological Association lower urinary tract symptom score >19). Testosterone replacement should not be administered to men with baseline hematocrit ≥50%, severe untreated obstructive sleep apnea, uncontrolled or poorly controlled congestive heart failure, or myocardial infarction, stroke, or acute coronary syndrome in the preceding 6 months.
CONDITIONS IN WHICH TESTOSTERONE ADMINISTRATION IS ASSOCIATED WITH A RISK OF ADVERSE OUTCOME |



