Learning Outcomes
After completing this chapter, you will be able to
 Define the study of biopharmaceutics.
Define the study of biopharmaceutics.
 List and describe the four major processes that make up the study of pharmacokinetics.
List and describe the four major processes that make up the study of pharmacokinetics.
 Describe factors that can alter the absorption of a medication.
Describe factors that can alter the absorption of a medication.
 List special precautions for the major classes of drugs.
List special precautions for the major classes of drugs.
 Describe how medications are distributed within the body, including factors that affect medication distribution in the body.
Describe how medications are distributed within the body, including factors that affect medication distribution in the body.
 List and describe the two most common types of drug interactions.
List and describe the two most common types of drug interactions.
 Define pharmacodynamics.
Define pharmacodynamics.
 Describe how medications are eliminated from the body, including factors (e.g., disease states) that can increase or decrease elimination of a medication.
Describe how medications are eliminated from the body, including factors (e.g., disease states) that can increase or decrease elimination of a medication.
 Describe the steps that must occur before a medication can exert its effect on the body.
Describe the steps that must occur before a medication can exert its effect on the body.
 Describe potential problems that can occur when a product formulation is disrupted or when absorption, distribution, metabolism, or elimination is altered, and how these alterations can affect the pharmacodynamics of a medication.
Describe potential problems that can occur when a product formulation is disrupted or when absorption, distribution, metabolism, or elimination is altered, and how these alterations can affect the pharmacodynamics of a medication.
Key Terms
| absorption | The amount of medication that enters the bloodstream, or systemic circulation. |
| bioavailability | The percentage of an administered dose of a medication that reaches the bloodstream. |
| biopharmaceutics | The study of the manufacture of medications for effective delivery into the body. It includes the relationships between the physical and chemical properties of a drug, the dosage form in which the drug is given, the route of administration, and the effects of properties and dosage on the rate and extent of drug absorption. |
| clearance | The removal of plasma which is completely cleared of drugs per unit of time. It combines the elimination rate with the flow of a drug through the organs of elimination (i.e., liver and kidneys). It is usually measured in mL/min or L/hr. |
| cytochrome P450 (CYP) | A group of enzymes that metabolize drugs. |
| disintegration | The breakdown of medication from its original solid formulation. |
Patient Variables Affecting Pharmacokinetics
|
This chapter describes the basic elements of biopharmaceutics, pharmacokinetics, and pharmacodynamics. These concepts are useful in describing how medications exert their effects on body systems, as well as how drugs affect body systems.
The pharmacy technician is often the first person in the pharmacy to handle a patient’s medication, often filling multiple prescriptions for a single patient. Thus, the pharmacy technician who prepares prescriptions for dispensing should have a basic understanding of biopharmaceutics, pharmacokinetics, and pharmacodynamics, which collectively describe how a particular medication is prepared, is handled by the body, and affects the body. Having this understanding can help avoid unwanted (adverse) drug events, because the complex interactions of many medications is a risk factor for adverse drug events—especially in older individuals and certain other patient populations, as will be described in more detail in this chapter.
For example, a prescriber may write that a particular prescription medication should be crushed before being taken; however, a knowledgeable pharmacy technician may know that crushing that particular medication results in loss of the slow-release properties of the drug. Likewise, a pharmacy technician knowledgeable about the absorption of a particular medication may know that it should not be taken with certain other medications or dietary agents that could affect its absorption.
An understanding of basic biopharmaceutical, pharmacokinetic, and pharmacodynamic principles can aid the clinician in appropriately monitoring medication therapy, and the pharmacy technician can aid the pharmacist in such surveillance.
Biopharmaceutics
Biopharmaceutics is the study of the manufacture of medications for effective delivery into the body. It includes the relationships between the physical and chemical properties of a drug, the dosage form in which the drug is given, the route of administration, and the effects of properties and dosage on the rate and extent of drug absorption. It has long been recognized that the method by which a medication is prepared and formulated can affect its action. The study of biopharmaceutics has led to the improved design of drug products to enhance the delivery of medication and ultimately to optimize the clinical effects of medication.
Medications can be formulated and administered to the patient in many different ways (see Chapter 12: Medication Dosage Forms and Routes of Administration). Common formulations include tablets, capsules, solutions, suspensions, suppositories, transdermal patches, creams, nasal sprays, and aerosolized medications. Routes of administration include oral, intraocular (in the eye), rectal, sublingual (under the tongue), buccal (cheek), topical, transdermal (through the skin), injectable, and inhaled.
Medications are formulated and administered differently for several reasons. In some case, a particular medication is available via multiple routes, and choosing one over the other may simply be a matter of preference or convenience. For example, the analgesic acetaminophen is available in many different dosage forms, including tablets, capsules, caplets, suspensions, and suppositories. For small children, administration by suspension or suppository will likely be easier than by a tablet. In other cases, different formulations and routes of administration are used because they can influence the rate, duration, or even extent of drug effect. Therefore, using the correct or optimal formulation is important and often has a bearing on the clinical effect of the medication.
Before most medications can exert a pharmacologic response, they need to be absorbed (taken up by the bloodstream), and before they can be absorbed, they need to be released from their formulations. A medication in a tablet formulation, for example, is more than just the active drug. Any given tablet will contain not only active drug but also non-drug ingredients, such as binders that keep the tablet from falling apart, fillers that add bulk to the tablet, and preservatives. Two processes—disintegration and dissolution, in that order—usually need to occur before the medication can be absorbed. Disintegration is the breakdown of the medication from its original solid formulation, and dissolution is the dissolving of medication into solution, usually in the stomach and intestinal tract. Disintegration aids dissolution, as smaller particles dissolve more easily than larger particles. Once a medication is disintegrated and dissolved, it can be absorbed into the bloodstream (figure 11-1). Some product formulations are disintegrated and dissolved more slowly or more rapidly than others, which may ultimately affect the rate of onset of therapeutic effect. For example, topical creams and ointments may be used to deliver drugs that are dissolved in special vehicles called organogels, such as pluronic lecithin organogel (PLO), which enhance the absorption of the drug across the skin (transdermal). Some injectable products can be designed to dissolve very slowly from the site of injection, such as intramuscular depot injections.
 Medications that are given intravenously have a very rapid onset of action because the medications are already in solution (therefore disintegration and dissolution do not need to occur) and are placed directly into the bloodstream.
Medications that are given intravenously have a very rapid onset of action because the medications are already in solution (therefore disintegration and dissolution do not need to occur) and are placed directly into the bloodstream.
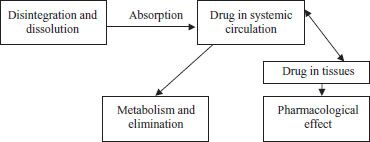
Figure 11–1. Relationship between drug and effect.
Pharmacokinetics
Pharmacokinetics is defined as the study of the movement of a drug through the body during the following phases: absorption, distribution, metabolism, and excretion. These processes are often referred to as “ADME.”
Absorption
Absorption refers to the amount of medication that enters the bloodstream, or systemic circulation. Not all the medication in a tablet, capsule, suppository, inhaler, or intramuscular or subcutaneous syringe enters the bloodstream, and therefore not all of it is absorbed. Except for topical products that are applied for a local effect, such as anti-inflammatory creams, only absorbed medication has the potential to exert a systemic pharmacologic effect.
The term bioavailability refers to the percentage of an administered dose of a medication that reaches the bloodstream. The bioavailability of an agent depends on many factors, including the amount of drug dissolved, its dosage form, and its route of administration.
Some medications that are ingested orally are metabolized (broken down) before they reach the main bloodstream, which is referred to as first-pass metabolism. This often results in a small percentage of the medication being metabolized on the first pass through either the intestine wall or the liver (figure 11-2). Medications that undergo a high first-pass metabolism have a high amount of drug metabolized, and therefore a lower percentage is available to reach the main systemic circulation.
There are some routes of drug administration, such as rectal, inhalation, and sublingual, that avoid first-pass metabolism because they bypass the small intestine and liver and the drugs are absorbed directly into the main bloodstream. For example, drugs such as ergotamine and nitroglycerin are better absorbed when given rectally and sublingually, respectively, than when given orally because if given orally, they are broken down in the small intestine and liver before they can be absorbed into the bloodstream.
Medications given intravenously are administered directly into the vein and therefore enter the bloodstream directly. Thus, they have 100% bioavailability, meaning that 100% of the dose administered is available in the bloodstream and has the potential to exert a pharmacologic effect.
 Usually, the dose needed orally is much more than the dose needed intravenously because the bioavailability of the oral formulation is much less than 100%.
Usually, the dose needed orally is much more than the dose needed intravenously because the bioavailability of the oral formulation is much less than 100%.
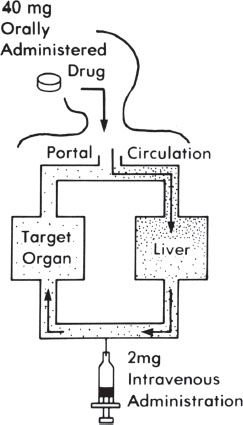
Figure 11–2. First-pass metabolism.
For example, levothyroxine, used to treat low thyroid levels, has an intravenous dose that is approximately one-half that of the oral dose, but will produce the same pharmacologic response. In other cases, the oral absorption of an agent is so poor that it must be given intravenously. Vancomycin is an example of an antibiotic that is normally used intravenously to treat serious infections because it is so poorly absorbed orally.
Distribution
Once absorbed into the bloodstream, medication either leaves the bloodstream and enters the tissues—including, but not limited to, the site of action (the tissue organ site where the medication is to exert its intended pharmacologic response)—or it remains in the blood, bound to protein components. Therefore, for any given dose of medication, some of it will travel to tissues and some will remain in the bloodstream. Some medications are highly bound to blood proteins, in some cases greater than 90%. The medication bound to blood proteins is inactive and does not exert any pharmacologic effect until it is released from the protein. Only medication that is free, or not bound to proteins, can leave the bloodstream and enter the tissues to exert a pharmacologic effect. Of the percentage that is free, or circulating to tissues, only the amount that reaches the target site will exert a pharmacologic effect (wanted or unwanted). This process is reversible; as drug is released from protein, it is distributed into tissues, leading to binding of drug to take its place on the protein (figure 11-3).
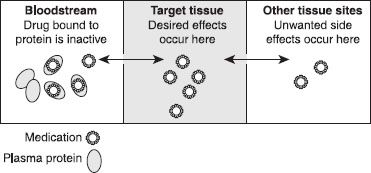
Figure 11–3. Medication distribution. This figure depicts the travel of medication from the bloodstream to the target tissue to other tissues. The double-headed arrows indicate that flow can occur in either direction. Medication bound to protein in the blood is inactive and cannot move (or distribute) to other tissues, but the medication can dissociate or separate from the protein. Once separated, it can move about the body.
Albumin is the main protein that binds medications in the blood. Some diseases, particularly those that damage the liver, can decrease the amount of albumin in the plasma. In such cases, medications that normally bind highly to albumin are unable to do so to the normal extent, and a greater proportion of those medications will be “free” and therefore active. Medications that are highly bound to albumin include phenytoin, which is used to treat seizures, and warfarin, an anticoagulant.
For some drugs, the amount of drug in the bloodstream, typically reported as the concentration of drug in blood (such as mg/L), can be measured in the clinical setting to help guide appropriate therapy. For example, after a drug is ingested orally, the concentration of drug in blood rises slowly, reaches a maximum amount, then drops slowly due to the drug being eliminated or metabolized (figure 11-4). Here, a range of concentrations of drug in the blood is achieved during a specified dosing regimen. For some drugs, the concentration may rise to a level that causes toxicity. These drugs often have side effects that appear with very small dose increments, and researchers have been able to determine the therapeutic level for these medications—the level at which most patients receive the desired effect with minimal side effects. Thus, blood levels of these medications can be measured to determine whether a change in therapy is needed on the basis of the laboratory results. Examples of medications whose levels are measured in the blood include many of the antiepileptic agents, such as phenytoin, carbamazepine, valproic acid, and phenobarbital; digoxin for atrial fibrillation; and some antibiotics, including gentamicin, tobramycin, and vancomycin.
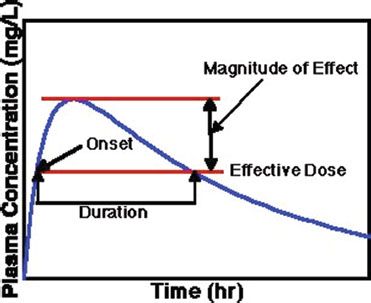
Figure 11–4. Illustration showing the relationship between drug concentration and drug effect, including therapeutic range.
The apparent volume of distribution for a drug describes the extent of its outreach to various tissues and spaces throughout the body. The space that the drug distributes into is often called a “compartment,” which is a hypothetical pool to which that drug is distributed. Most medications are distributed throughout the bloodstream and in many organs, fluids, and tissues (fat tissue or lean muscle tissue); other medications are not as widely distributed. In general, medications with a large volume of distribution will have a lower blood concentration, whereas medications with a small volume of distribution will have a higher blood concentration (figure 11-5). A simple way to think of this is to picture a bucket of water. A certain amount of dissolvable red food coloring placed into the bucket will be spread to all areas of the bucket. The same amount of coloring placed in a bucket one-half the size of the first will also spread to all areas of the bucket, but will look more colorful (darker red) because the water volume is smaller. The smaller bucket will appear to contain twice the amount of red color, when actually the same amount of coloring was used in both cases.
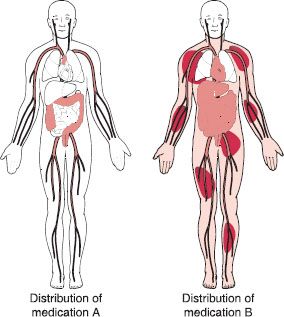
Figure 11–5. Volume of distribution. This schematic hypothetically and simplistically depicts drug distribution throughout the body. Medication A, shown on the left, appears to remain primarily in the bloodstream and is distributed to the target organ, the large intestines. Medication B, shown on the right, appears to be widely distributed throughout the body.
Certain other factors can also affect the extent of distribution of a drug throughout the body. Some medications that are highly bound (attracted) to proteins found in the bloodstream often have low volumes of distribution because they tend to stay in the bloodstream. These medications often have very high blood concentrations, and the amount (dose) of medication is relatively small compared with other drugs. Conversely, medications that have a high affinity to body fat and medications that either are bound to proteins in tissue or are not highly bound to plasma proteins tend to have high volumes of distribution—that is, they tend to be widely distributed throughout the body. The doses of these medications can be very high, sometimes upwards of 1–2 grams per day.
How does knowing the volume of distribution of a medication help with drug administration? Knowing the volume of distribution of a certain medication can help the prescriber to approximate the dose of medication needed to attain the desired level of drug in the body for it to be effective or to start working quickly. If a medication is widely distributed through the body and the prescriber wants the medication to start working quickly, sometimes a loading dose of the medication (a larger first dose) will be given to more quickly achieve a higher drug concentration in the body. Examples of medications for which a loading dose is sometimes given include phenytoin, digoxin, and some antibiotics.
As a general example, a typical prescription for the antibiotic azithromycin may be written as follows: “Take 2 tablets today (500 mg), then take 1 tablet daily (250 mg) for 4 days.” Two tablets are used the first day as a loading dose, to help achieve a certain blood level of the medication to jump-start therapy. If a medication is not widely distributed through the body, a smaller loading dose or no loading dose will be needed. Most medications do not require a loading dose.
Metabolism
The breakdown and elimination of drugs from the body occurs by metabolism and excretion. The metabolism of medication is the breakdown of medication in the body. Here, the drug molecule is changed or altered in some way to create a secondary molecule called a metabolite. Some drug molecules are not susceptible to metabolic breakdown, and may travel directly to the kidneys to be excreted (explained in more detail below). The liver is the major organ in which drug metabolism occurs, although significant metabolism can occur in the small intestine. Very little metabolism occurs in other organs, such as the kidneys and lungs. Metabolism is most often accomplished by protein substances called enzymes. The most common enzymes that metabolize drugs belong to a family of enzymes called the cytochrome P450 (CYP) system. These enzymes are very active in converting a drug to its metabolites.
In most cases, metabolism of a drug results in the formation of metabolites (altered chemical forms of the drug) that are not pharmacologically active in the human body. This results in a drop in the concentration of active drug in the body, and usually leads to a loss of drug activity because the metabolites are not binding to the drug receptors. However, in rare cases, metabolism can result in the formation of an active metabolite that may be more pharmacologically active than the parent (original) drug, or even toxic. It is therefore important to know whether the active metabolite acts in the same manner as the parent drug. There are also a few examples where a drug is administered in an inactive form, which is metabolized or converted to the active component. This inactive form is called a “pro-drug,” and examples include enalapril and valacyclovir.
An example of an active metabolite is seen with the drug fluoxetine, used primarily for depression. Fluoxetine is metabolized to norfluoxetine, which is active and exerts an antidepressant effect that is even longeracting than fluoxetine. This prolonged effect can be good or bad, depending on the situation and specific patient characteristics. An advantage is that a long-acting metabolite allows convenient dosing of the medication once a day, or less frequently with the advent of a onceweekly formulation. A disadvantage is that this longacting metabolite can remain in the system longer than wanted, so any side effects may occur for a prolonged period, especially in the elderly, who tend to eliminate the drug more slowly.
Excretion
Excretion refers to the irreversible removal of a drug or metabolite from a body fluid. The most common location of drug excretion in the body is the kidneys, with the biliary tract being another important route of excretion. In the kidney, some medications are excreted from the blood by a filtering process whereby drug is eliminated into the urine without being metabolized. After drug metabolism, many metabolites are water soluble, making them very susceptible to excretion by the kidneys. In fact, studies are often conducted to measure the recovery of intact drug and metabolites in urine, to understand the pharmacokinetics of new drugs. Drugs and metabolites may also be excreted from the liver into the bile. Here, drug is transferred from the bloodstream into the liver cell, then into the bile duct. From here, the drug travels through the bile duct and into the small intestine for elimination.
The total removal of a drug via metabolism and/or excretion from the blood stream per unit of time can also be referred to as drug clearance, which combines elimination rate with the flow of a drug through the organs of elimination (i.e., liver and kidneys). This term is often used to compare various drug pharmacokinetics based on organ blood flow values (in units of mL/min or L/hr).
The half-life, often designated by the term “T1/2,” of a drug is also related to clearance and elimination. Here, half-life refers to the time that it takes for 50% of an amount of drug to be eliminated from the body. A drug with low clearance, meaning one that is not effectively metabolized or excreted, will typically have a long halflife. This means that it will take a long time to eliminate a dose of the drug from the body. Here, drug administration that is too rapid (faster than the drug can be eliminated) can lead to excessive accumulation of the drug in the body. Such drugs have a much higher risk of toxicity than drugs with a short half-life and high clearance.
Drug Interactions
Drugs (and some foods) can compete with other drugs for metabolism within the system or alter the metabolic system altogether, resulting in drug interactions. A drug interaction is defined as the impact of a drug or food product on the amount or activity of another drug in the body. This drug-drug (or drug-food) interaction can result in enhanced, reduced, or new activity of the drug in the body. The most common cause of drug interactions is altered drug metabolism in the liver. For example, some medications, foods, and herbal products can inhibit (or slow down) enzyme activity, which results in reduced drug metabolism of other medications. Conversely, some drugs can induce (or speed up) the metabolism of other medications. These two mechanisms are the most common forms of drug interactions. Thus, knowing the metabolic effects of each medication dispensed to a patient can help determine if a drug interaction is likely to exist.
For example, some antiepileptic medications are referred to as enzyme inducers, meaning they up-regulate the metabolism of many other medications. Phenytoin, an antiepileptic agent, is an enzyme inducer, and it is known to increase the metabolism of more than 100 other medications. This can result in lessening the activity of the other medications, because they are broken down and excreted from the body more quickly than normal. Conversely, some medications can inhibit the metabolism of other medications. For example, erythromycin, some antifungal medications, and cyclosporine are all potent enzyme inhibitors, or medications that are known to reduce the metabolism of many other medications. This can result in higher, more prolonged blood levels of these other medications, which can lead to prolonged duration of action of the other medications and the development of side effects. An example of a drug-drug interaction is that of erythromycin (a CYP enzyme inhibitor) with the bronchodilator theophylline (metabolized by CYP), resulting in systemic accumulation of theophylline and toxicity. An example of a drug-food interaction is ingestion of grapefruit juice (a CYP enzyme inhibitor) with the antihypertensive nifedipine (metabolized by CYP), resulting in hypotensive episodes.
Patient Variables Affecting Pharmacokinetics
Several factors can affect the normal processes of ADME, leading to altered pharmacokinetics. For example, drug absorption can be altered by a change in the speed of the gastrointestinal tract, such as constipation or diarrhea. The elimination or clearance of a drug can be impacted by diseases of the kidney and liver, such as cirrhosis. Reduced elimination can then lead to a prolonged halflife of the drug when compared with patients without organ dysfunction. Changes in heart function or cardiac output can lead to changes in delivery of drugs via the bloodstream. Severe heart failure, in which there is low cardiac output, can lead to decreased blood flow to the kidneys and liver and therefore to decreased clearance of medications.
 Patients who are markedly overweight or underweight may have differing abilities to eliminate medications because kidney and liver size and blood flow are proportional to body weight.
Patients who are markedly overweight or underweight may have differing abilities to eliminate medications because kidney and liver size and blood flow are proportional to body weight.



