PART 2: Cardinal Manifestations and Presentation of Diseases
SECTION 1 PAIN
18 | Pain: Pathophysiology and Management |
The province of medicine is to preserve and restore health and to relieve suffering. Understanding pain is essential to both of these goals. Because pain is universally understood as a signal of disease, it is the most common symptom that brings a patient to a physician’s attention. The function of the pain sensory system is to protect the body and maintain homeostasis. It does this by detecting, localizing, and identifying potential or actual tissue-damaging processes. Because different diseases produce characteristic patterns of tissue damage, the quality, time course, and location of a patient’s pain lend important diagnostic clues. It is the physician’s responsibility to provide rapid and effective pain relief.
THE PAIN SENSORY SYSTEM
Pain is an unpleasant sensation localized to a part of the body. It is often described in terms of a penetrating or tissue-destructive process (e.g., stabbing, burning, twisting, tearing, squeezing) and/or of a bodily or emotional reaction (e.g., terrifying, nauseating, sickening). Furthermore, any pain of moderate or higher intensity is accompanied by anxiety and the urge to escape or terminate the feeling. These properties illustrate the duality of pain: it is both sensation and emotion. When it is acute, pain is characteristically associated with behavioral arousal and a stress response consisting of increased blood pressure, heart rate, pupil diameter, and plasma cortisol levels. In addition, local muscle contraction (e.g., limb flexion, abdominal wall rigidity) is often present.
PERIPHERAL MECHANISMS
The Primary Afferent Nociceptor A peripheral nerve consists of the axons of three different types of neurons: primary sensory afferents, motor neurons, and sympathetic postganglionic neurons (Fig. 18-1). The cell bodies of primary sensory afferents are located in the dorsal root ganglia within the vertebral foramina. The primary afferent axon has two branches: one projects centrally into the spinal cord and the other projects peripherally to innervate tissues. Primary afferents are classified by their diameter, degree of myelination, and conduction velocity. The largest diameter afferent fibers, A-beta (Aβ), respond maximally to light touch and/or moving stimuli; they are present primarily in nerves that innervate the skin. In normal individuals, the activity of these fibers does not produce pain. There are two other classes of primary afferent nerve fibers: the small diameter myelinated A-delta (Aδ) and the unmyelinated (C) axons (Fig. 18-1). These fibers are present in nerves to the skin and to deep somatic and visceral structures. Some tissues, such as the cornea, are innervated only by Aδ and C fiber afferents. Most Aδ and C fiber afferents respond maximally only to intense (painful) stimuli and produce the subjective experience of pain when they are electrically stimulated; this defines them as primary afferent nociceptors (pain receptors). The ability to detect painful stimuli is completely abolished when conduction in Aδ and C fiber axons is blocked.
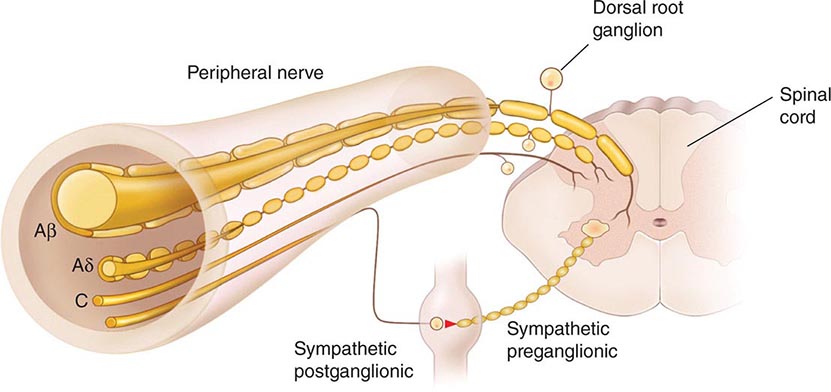
FIGURE 18-1 Components of a typical cutaneous nerve. There are two distinct functional categories of axons: primary afferents with cell bodies in the dorsal root ganglion, and sympathetic postganglionic fibers with cell bodies in the sympathetic ganglion. Primary afferents include those with large-diameter myelinated (Aβ), small-diameter myelinated (Aδ), and unmyelinated (C) axons. All sympathetic postganglionic fibers are unmyelinated.
Individual primary afferent nociceptors can respond to several different types of noxious stimuli. For example, most nociceptors respond to heat; intense cold; intense mechanical distortion, such as a pinch; changes in pH, particularly an acidic environment; and application of chemical irritants including adenosine triphosphate (ATP), serotonin, bradykinin, and histamine.
Sensitization When intense, repeated, or prolonged stimuli are applied to damaged or inflamed tissues, the threshold for activating primary afferent nociceptors is lowered, and the frequency of firing is higher for all stimulus intensities. Inflammatory mediators such as bradykinin, nerve-growth factor, some prostaglandins, and leukotrienes contribute to this process, which is called sensitization. Sensitization occurs at the level of the peripheral nerve terminal (peripheral sensitization) as well as at the level of the dorsal horn of the spinal cord (central sensitization). Peripheral sensitization occurs in damaged or inflamed tissues, when inflammatory mediators activate intracellular signal transduction in nociceptors, prompting an increase in the production, transport, and membrane insertion of chemically gated and voltage-gated ion channels. These changes increase the excitability of nociceptor terminals and lower their threshold for activation by mechanical, thermal, and chemical stimuli. Central sensitization occurs when activity, generated by nociceptors during inflammation, enhances the excitability of nerve cells in the dorsal horn of the spinal cord. Following injury and resultant sensitization, normally innocuous stimuli can produce pain (termed allodynia). Sensitization is a clinically important process that contributes to tenderness, soreness, and hyperalgesia (increased pain intensity in response to the same noxious stimulus; e.g., moderate pressure causes severe pain). A striking example of sensitization is sunburned skin, in which severe pain can be produced by a gentle slap on the back or a warm shower.
Sensitization is of particular importance for pain and tenderness in deep tissues. Viscera are normally relatively insensitive to noxious mechanical and thermal stimuli, although hollow viscera do generate significant discomfort when distended. In contrast, when affected by a disease process with an inflammatory component, deep structures such as joints or hollow viscera characteristically become exquisitely sensitive to mechanical stimulation.
A large proportion of Aδ and C fiber afferents innervating viscera are completely insensitive in normal noninjured, noninflamed tissue. That is, they cannot be activated by known mechanical or thermal stimuli and are not spontaneously active. However, in the presence of inflammatory mediators, these afferents become sensitive to mechanical stimuli. Such afferents have been termed silent nociceptors, and their characteristic properties may explain how, under pathologic conditions, the relatively insensitive deep structures can become the source of severe and debilitating pain and tenderness. Low pH, prostaglandins, leukotrienes, and other inflammatory mediators such as bradykinin play a significant role in sensitization.
Nociceptor-Induced Inflammation Primary afferent nociceptors also have a neuroeffector function. Most nociceptors contain polypeptide mediators that are released from their peripheral terminals when they are activated (Fig. 18-2). An example is substance P, an 11-amino-acid peptide. Substance P is released from primary afferent nociceptors and has multiple biologic activities. It is a potent vasodilator, degranulates mast cells, is a chemoattractant for leukocytes, and increases the production and release of inflammatory mediators. Interestingly, depletion of substance P from joints reduces the severity of experimental arthritis. Primary afferent nociceptors are not simply passive messengers of threats to tissue injury but also play an active role in tissue protection through these neuroeffector functions.
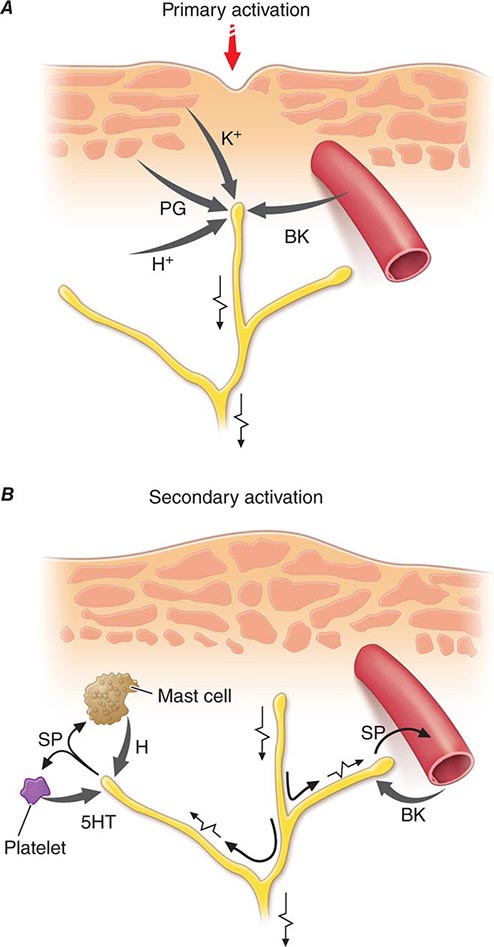
FIGURE 18-2 Events leading to activation, sensitization, and spread of sensitization of primary afferent nociceptor terminals. A. Direct activation by intense pressure and consequent cell damage. Cell damage induces lower pH (H+) and leads to release of potassium (K+) and to synthesis of prostaglandins (PG) and bradykinin (BK). Prostaglandins increase the sensitivity of the terminal to bradykinin and other pain-producing substances. B. Secondary activation. Impulses generated in the stimulated terminal propagate not only to the spinal cord but also into other terminal branches where they induce the release of peptides, including substance P (SP). Substance P causes vasodilation and neurogenic edema with further accumulation of bradykinin (BK). Substance P also causes the release of histamine (H) from mast cells and serotonin (5HT) from platelets.
CENTRAL MECHANISMS
The Spinal Cord and Referred Pain The axons of primary afferent nociceptors enter the spinal cord via the dorsal root. They terminate in the dorsal horn of the spinal gray matter (Fig. 18-3). The terminals of primary afferent axons contact spinal neurons that transmit the pain signal to brain sites involved in pain perception. When primary afferents are activated by noxious stimuli, they release neurotransmitters from their terminals that excite the spinal cord neurons. The major neurotransmitter released is glutamate, which rapidly excites dorsal horn neurons. Primary afferent nociceptor terminals also release peptides, including substance P and calcitonin gene-related peptide, which produce a slower and longer-lasting excitation of the dorsal horn neurons. The axon of each primary afferent contacts many spinal neurons, and each spinal neuron receives convergent inputs from many primary afferents.
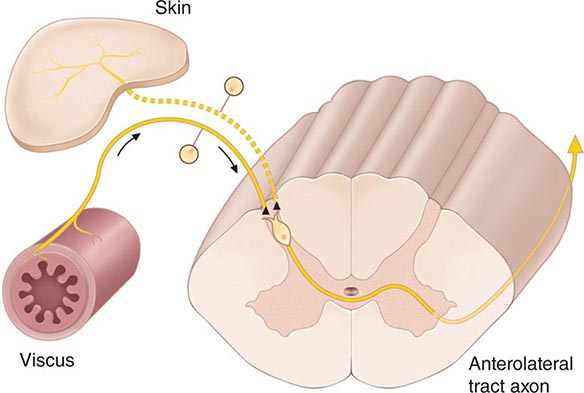
FIGURE 18-3 The convergence-projection hypothesis of referred pain. According to this hypothesis, visceral afferent nociceptors converge on the same pain-projection neurons as the afferents from the somatic structures in which the pain is perceived. The brain has no way of knowing the actual source of input and mistakenly “projects” the sensation to the somatic structure.
The convergence of sensory inputs to a single spinal pain-transmission neuron is of great importance because it underlies the phenomenon of referred pain. All spinal neurons that receive input from the viscera and deep musculoskeletal structures also receive input from the skin. The convergence patterns are determined by the spinal segment of the dorsal root ganglion that supplies the afferent innervation of a structure. For example, the afferents that supply the central diaphragm are derived from the third and fourth cervical dorsal root ganglia. Primary afferents with cell bodies in these same ganglia supply the skin of the shoulder and lower neck. Thus, sensory inputs from both the shoulder skin and the central diaphragm converge on pain-transmission neurons in the third and fourth cervical spinal segments. Because of this convergence and the fact that the spinal neurons are most often activated by inputs from the skin, activity evoked in spinal neurons by input from deep structures is mislocalized by the patient to a place that roughly corresponds with the region of skin innervated by the same spinal segment. Thus, inflammation near the central diaphragm is often reported as shoulder discomfort. This spatial displacement of pain sensation from the site of the injury that produces it is known as referred pain.
Ascending Pathways for Pain A majority of spinal neurons contacted by primary afferent nociceptors send their axons to the contralateral thalamus. These axons form the contralateral spinothalamic tract, which lies in the anterolateral white matter of the spinal cord, the lateral edge of the medulla, and the lateral pons and midbrain. The spinothalamic pathway is crucial for pain sensation in humans. Interruption of this pathway produces permanent deficits in pain and temperature discrimination.
Spinothalamic tract axons ascend to several regions of the thalamus. There is tremendous divergence of the pain signal from these thalamic sites to several distinct areas of the cerebral cortex that subserve different aspects of the pain experience (Fig. 18-4). One of the thalamic projections is to the somatosensory cortex. This projection mediates the purely sensory aspects of pain, i.e., its location, intensity, and quality. Other thalamic neurons project to cortical regions that are linked to emotional responses, such as the cingulate gyrus and other areas of the frontal lobes, including the insular cortex. These pathways to the frontal cortex subserve the affective or unpleasant emotional dimension of pain. This affective dimension of pain produces suffering and exerts potent control of behavior. Because of this dimension, fear is a constant companion of pain. As a consequence, injury or surgical lesions to areas of the frontal cortex activated by painful stimuli can diminish the emotional impact of pain while largely preserving the individual’s ability to recognize noxious stimuli as painful.
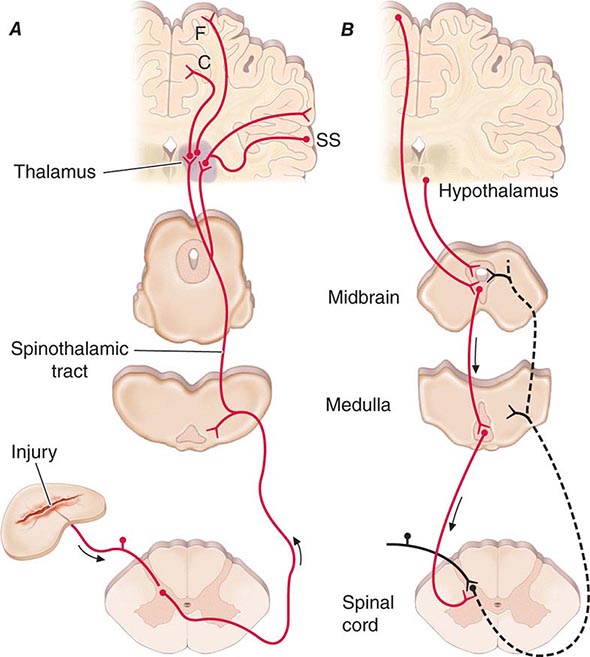
FIGURE 18-4 Pain transmission and modulatory pathways. A. Transmission system for nociceptive messages. Noxious stimuli activate the sensitive peripheral ending of the primary afferent nociceptor by the process of transduction. The message is then transmitted over the peripheral nerve to the spinal cord, where it synapses with cells of origin of the major ascending pain pathway, the spinothalamic tract. The message is relayed in the thalamus to the anterior cingulate (C), frontal insular (F), and somatosensory cortex (SS). B. Pain-modulation network. Inputs from frontal cortex and hypothalamus activate cells in the midbrain that control spinal pain-transmission cells via cells in the medulla.
PAIN MODULATION
The pain produced by injuries of similar magnitude is remarkably variable in different situations and in different individuals. For example, athletes have been known to sustain serious fractures with only minor pain, and Beecher’s classic World War II survey revealed that many soldiers in battle were unbothered by injuries that would have produced agonizing pain in civilian patients. Furthermore, even the suggestion that a treatment will relieve pain can have a significant analgesic effect (the placebo effect). On the other hand, many patients find even minor injuries (such as venipuncture) frightening and unbearable, and the expectation of pain can induce pain even without a noxious stimulus. The suggestion that pain will worsen following administration of an inert substance can increase its perceived intensity (the nocebo effect).
The powerful effect of expectation and other psychological variables on the perceived intensity of pain is explained by brain circuits that modulate the activity of the pain-transmission pathways. One of these circuits has links to the hypothalamus, midbrain, and medulla, and it selectively controls spinal pain-transmission neurons through a descending pathway (Fig. 18-4).
Human brain–imaging studies have implicated this pain-modulating circuit in the pain-relieving effect of attention, suggestion, and opioid analgesic medications (Fig. 18-5). Furthermore, each of the component structures of the pathway contains opioid receptors and is sensitive to the direct application of opioid drugs. In animals, lesions of this descending modulatory system reduce the analgesic effect of systemically administered opioids such as morphine. Along with the opioid receptor, the component nuclei of this pain-modulating circuit contain endogenous opioid peptides such as the enkephalins and β-endorphin.
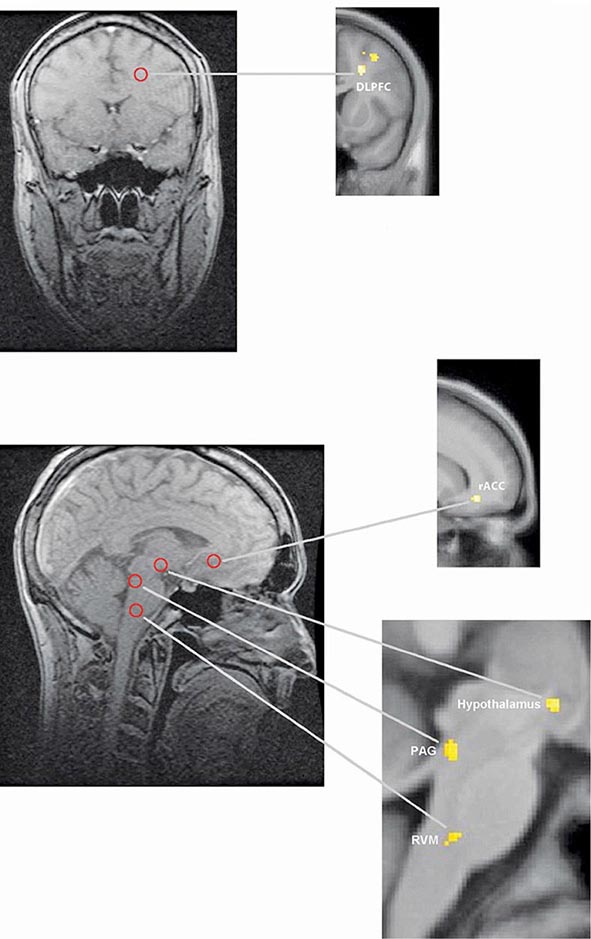
FIGURE 18-5 Functional magnetic resonance imaging (fMRI) demonstrates placebo-enhanced brain activity in anatomic regions correlating with the opioidergic descending pain control system. Top panel: Frontal fMRI image shows placebo-enhanced brain activity in the dorsal lateral prefrontal cortex (DLPFC). Bottom panel: Sagittal fMRI images show placebo-enhanced responses in the rostral anterior cingulate cortex (rACC), the rostral ventral medullae (RVM), the periaqueductal gray (PAG) area, and the hypothalamus. The placebo-enhanced activity in all areas was reduced by naloxone, demonstrating the link between the descending opioidergic system and the placebo analgesic response. (Adapted with permission from F Eippert et al: Neuron 63:533, 2009.)
The most reliable way to activate this endogenous opioid-mediated modulating system is by suggestion of pain relief or by intense emotion directed away from the pain-causing injury (e.g., during severe threat or an athletic competition). In fact, pain-relieving endogenous opioids are released following surgical procedures and in patients given a placebo for pain relief.
Pain-modulating circuits can enhance as well as suppress pain. Both pain-inhibiting and pain-facilitating neurons in the medulla project to and control spinal pain-transmission neurons. Because pain-transmission neurons can be activated by modulatory neurons, it is theoretically possible to generate a pain signal with no peripheral noxious stimulus. In fact, human functional imaging studies have demonstrated increased activity in this circuit during migraine headaches. A central circuit that facilitates pain could account for the finding that pain can be induced by suggestion or enhanced by expectation and provides a framework for understanding how psychological factors can contribute to chronic pain.
NEUROPATHIC PAIN
Lesions of the peripheral or central nociceptive pathways typically result in a loss or impairment of pain sensation. Paradoxically, damage to or dysfunction of these pathways can also produce pain. For example, damage to peripheral nerves, as occurs in diabetic neuropathy, or to primary afferents, as in herpes zoster infection, can result in pain that is referred to the body region innervated by the damaged nerves. Pain may also be produced by damage to the central nervous system (CNS), for example, in some patients following trauma or vascular injury to the spinal cord, brainstem, or thalamic areas that contain central nociceptive pathways. Such neuropathic pains are often severe and are typically resistant to standard treatments for pain.
Neuropathic pain typically has an unusual burning, tingling, or electric shock–like quality and may be triggered by very light touch. These features are rare in other types of pain. On examination, a sensory deficit is characteristically present in the area of the patient’s pain. Hyperpathia, a greatly exaggerated pain sensation to innocuous or mild nociceptive stimuli, is also characteristic of neuropathic pain; patients often complain that the very lightest moving stimulus evokes exquisite pain (allodynia). In this regard, it is of clinical interest that a topical preparation of 5% lidocaine in patch form is effective for patients with postherpetic neuralgia who have prominent allodynia.
A variety of mechanisms contribute to neuropathic pain. As with sensitized primary afferent nociceptors, damaged primary afferents, including nociceptors, become highly sensitive to mechanical stimulation and may generate impulses in the absence of stimulation. Increased sensitivity and spontaneous activity are due, in part, to an increased concentration of sodium channels in the damaged nerve fiber. Damaged primary afferents may also develop sensitivity to norepinephrine. Interestingly, spinal cord pain-transmission neurons cut off from their normal input may also become spontaneously active. Thus, both CNS and peripheral nervous system hyperactivity contribute to neuropathic pain.
Sympathetically Maintained Pain Patients with peripheral nerve injury occasionally develop spontaneous pain in the region innervated by the nerve. This pain is often described as having a burning quality. The pain typically begins after a delay of hours to days or even weeks and is accompanied by swelling of the extremity, periarticular bone loss, and arthritic changes in the distal joints. The pain may be relieved by a local anesthetic block of the sympathetic innervation to the affected extremity. Damaged primary afferent nociceptors acquire adrenergic sensitivity and can be activated by stimulation of the sympathetic outflow. This constellation of spontaneous pain and signs of sympathetic dysfunction following injury has been termed complex regional pain syndrome (CRPS). When this occurs after an identifiable nerve injury, it is termed CRPS type II (also known as posttraumatic neuralgia or, if severe, causalgia). When a similar clinical picture appears without obvious nerve injury, it is termed CRPS type I (also known as reflex sympathetic dystrophy). CRPS can be produced by a variety of injuries, including fractures of bone, soft tissue trauma, myocardial infarction, and stroke (Chap. 446). CRPS type I typically resolves with symptomatic treatment; however, when it persists, detailed examination often reveals evidence of peripheral nerve injury. Although the pathophysiology of CRPS is poorly understood, the pain and the signs of inflammation, when acute, can be rapidly relieved by blocking the sympathetic nervous system. This implies that sympathetic activity can activate undamaged nociceptors when inflammation is present. Signs of sympathetic hyperactivity should be sought in patients with posttraumatic pain and inflammation and no other obvious explanation.
TREATMENTACUTE PAIN
The ideal treatment for any pain is to remove the cause; thus, while treatment can be initiated immediately, efforts to establish the underlying etiology should always proceed as treatment begins. Sometimes, treating the underlying condition does not immediately relieve pain. Furthermore, some conditions are so painful that rapid and effective analgesia is essential (e.g., the postoperative state, burns, trauma, cancer, or sickle cell crisis). Analgesic medications are a first line of treatment in these cases, and all practitioners should be familiar with their use.
ASPIRIN, ACETAMINOPHEN, AND NONSTEROIDAL ANTI-INFLAMMATORY AGENTS (NSAIDs)
These drugs are considered together because they are used for similar problems and may have a similar mechanism of action (Table 18-1). All these compounds inhibit cyclooxygenase (COX), and, except for acetaminophen, all have anti-inflammatory actions, especially at higher dosages. They are particularly effective for mild to moderate headache and for pain of musculoskeletal origin.
DRUGS FOR RELIEF OF PAIN |
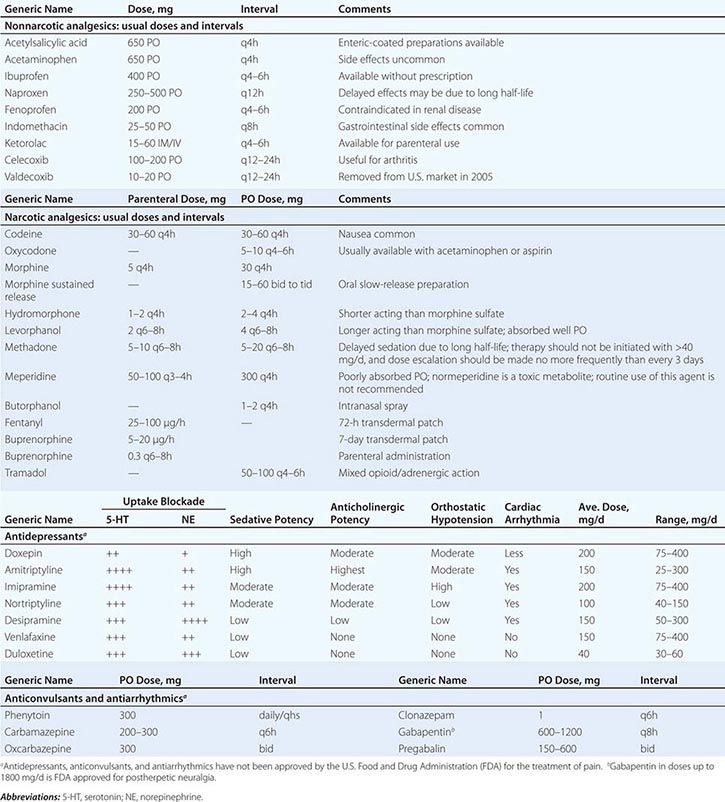
Because they are effective for these common types of pain and are available without prescription, COX inhibitors are by far the most commonly used analgesics. They are absorbed well from the gastrointestinal tract and, with occasional use, have only minimal side effects. With chronic use, gastric irritation is a common side effect of aspirin and NSAIDs and is the problem that most frequently limits the dose that can be given. Gastric irritation is most severe with aspirin, which may cause erosion and ulceration of the gastric mucosa leading to bleeding or perforation. Because aspirin irreversibly acetylates platelet cyclooxygenase and thereby interferes with coagulation of the blood, gastrointestinal bleeding is a particular risk. Older age and history of gastrointestinal disease increase the risks of aspirin and NSAIDs. In addition to the well-known gastrointestinal toxicity of NSAIDs, nephrotoxicity is a significant problem for patients using these drugs on a chronic basis. Patients at risk for renal insufficiency, particularly those with significant contraction of their intravascular volume as occurs with chronic diuretic use or acute hypovolemia, should be monitored closely. NSAIDs can also increase blood pressure in some individuals. Long-term treatment with NSAIDs requires regular blood pressure monitoring and treatment if necessary. Although toxic to the liver when taken in high doses, acetaminophen rarely produces gastric irritation and does not interfere with platelet function.
The introduction of parenteral forms of NSAIDs, ketorolac and diclofenac, extends the usefulness of this class of compounds in the management of acute severe pain. Both agents are sufficiently potent and rapid in onset to supplant opioids for many patients with acute severe headache and musculoskeletal pain.
There are two major classes of COX: COX-1 is constitutively expressed, and COX-2 is induced in the inflammatory state. COX-2–selective drugs have similar analgesic potency and produce less gastric irritation than the nonselective COX inhibitors. The use of COX-2–selective drugs does not appear to lower the risk of nephrotoxicity compared to nonselective NSAIDs. On the other hand, COX-2–selective drugs offer a significant benefit in the management of acute postoperative pain because they do not affect blood coagulation. Nonselective COX inhibitors are usually contraindicated postoperatively because they impair platelet-mediated blood clotting and are thus associated with increased bleeding at the operative site. COX-2 inhibitors, including celecoxib (Celebrex), are associated with increased cardiovascular risk. It appears that this is a class effect of NSAIDs, excluding aspirin. These drugs are contraindicated in patients in the immediate period after coronary artery bypass surgery and should be used with caution in elderly patients and those with a history of or significant risk factors for cardiovascular disease.
OPIOID ANALGESICS
Opioids are the most potent pain-relieving drugs currently available. Of all analgesics, they have the broadest range of efficacy and provide the most reliable and effective method for rapid pain relief. Although side effects are common, most are reversible: nausea, vomiting, pruritus, and constipation are the most frequent and bothersome side effects. Respiratory depression is uncommon at standard analgesic doses, but can be life-threatening. Opioid-related side effects can be reversed rapidly with the narcotic antagonist naloxone. Many physicians, nurses, and patients have a certain trepidation about using opioids that is based on an exaggerated fear of addiction. In fact, there is a vanishingly small chance of patients becoming addicted to narcotics as a result of their appropriate medical use. The physician should not hesitate to use opioid analgesics in patients with acute severe pain. Table 18-1 lists the most commonly used opioid analgesics.
Opioids produce analgesia by actions in the CNS. They activate pain-inhibitory neurons and directly inhibit pain-transmission neurons. Most of the commercially available opioid analgesics act at the same opioid receptor (μ-receptor), differing mainly in potency, speed of onset, duration of action, and optimal route of administration. Some side effects are due to accumulation of nonopioid metabolites that are unique to individual drugs. One striking example of this is normeperidine, a metabolite of meperidine. At higher doses of meperidine, typically greater than 1 g/d, accumulation of normeperidine can produce hyperexcitability and seizures that are not reversible with naloxone. Normeperidine accumulation is increased in patients with renal failure.
The most rapid pain relief is obtained by intravenous administration of opioids; relief with oral administration is significantly slower. Because of the potential for respiratory depression, patients with any form of respiratory compromise must be kept under close observation following opioid administration; an oxygen-saturation monitor may be useful, but only in a setting where the monitor is under constant surveillance. Opioid-induced respiratory depression is typically accompanied by sedation and a reduction in respiratory rate. A fall in oxygen saturation represents a critical level of respiratory depression and the need for immediate intervention to prevent life-threatening hypoxemia. Ventilatory assistance should be maintained until the opioid-induced respiratory depression has resolved. The opioid antagonist naloxone should be readily available whenever opioids are used at high doses or in patients with compromised pulmonary function. Opioid effects are dose-related, and there is great variability among patients in the doses that relieve pain and produce side effects. Synergistic respiratory depression is common when opioids are administered with other CNS depressants, most commonly the benzodiazepines. Because of this, initiation of therapy requires titration to optimal dose and interval. The most important principle is to provide adequate pain relief. This requires determining whether the drug has adequately relieved the pain and frequent reassessment to determine the optimal interval for dosing. The most common error made by physicians in managing severe pain with opioids is to prescribe an inadequate dose. Because many patients are reluctant to complain, this practice leads to needless suffering. In the absence of sedation at the expected time of peak effect, a physician should not hesitate to repeat the initial dose to achieve satisfactory pain relief.
An innovative approach to the problem of achieving adequate pain relief is the use of patient-controlled analgesia (PCA). PCA uses a microprocessor-controlled infusion device that can deliver a baseline continuous dose of an opioid drug as well as preprogrammed additional doses whenever the patient pushes a button. The patient can then titrate the dose to the optimal level. This approach is used most extensively for the management of postoperative pain, but there is no reason why it should not be used for any hospitalized patient with persistent severe pain. PCA is also used for short-term home care of patients with intractable pain, such as that caused by metastatic cancer.
It is important to understand that the PCA device delivers small, repeated doses to maintain pain relief; in patients with severe pain, the pain must first be brought under control with a loading dose before transitioning to the PCA device. The bolus dose of the drug (typically 1 mg of morphine, 0.2 mg of hydromorphone, or 10 μg of fentanyl) can then be delivered repeatedly as needed. To prevent overdosing, PCA devices are programmed with a lockout period after each demand dose is delivered (5–10 min) and a limit on the total dose delivered per hour. Although some have advocated the use of a simultaneous continuous or basal infusion of the PCA drug, this increases the risk of respiratory depression and has not been shown to increase the overall efficacy of the technique.
The availability of new routes of administration has extended the usefulness of opioid analgesics. Most important is the availability of spinal administration. Opioids can be infused through a spinal catheter placed either intrathecally or epidurally. By applying opioids directly to the spinal or epidural space adjacent to the spinal cord, regional analgesia can be obtained using relatively low total doses. Indeed, the dose required to produce effective localized analgesia when using morphine intrathecally (0.1–0.3 mg) is a fraction of that required to produce similar analgesia when administered intravenously (5–10 mg). In this way, side effects such as sedation, nausea, and respiratory depression can be minimized. This approach has been used extensively during labor and delivery and for postoperative pain relief following surgical procedures. Continuous intrathecal delivery via implanted spinal drug-delivery systems is now commonly used, particularly for the treatment of cancer-related pain that would require sedating doses for adequate pain control if given systemically. Opioids can also be given intranasally (butorphanol), rectally, and transdermally (fentanyl and buprenorphine), or through the oral mucosa (fentanyl), thus avoiding the discomfort of frequent injections in patients who cannot be given oral medication. The fentanyl and buprenorphine transdermal patches have the advantage of providing fairly steady plasma levels, which maximizes patient comfort.
Recent additions to the armamentarium for treating opioid-induced side effects are the peripherally acting opioid antagonists alvimopan (Entereg) and methylnaltrexone (Rellistor). Alvimopan is available as an orally administered agent that is restricted to the intestinal lumen by limited absorption; methylnaltrexone is available in a subcutaneously administered form that has virtually no penetration into the CNS. Both agents act by binding to peripheral μ-receptors, thereby inhibiting or reversing the effects of opioids at these peripheral sites. The action of both agents is restricted to receptor sites outside of the CNS; thus, these drugs can reverse the adverse effects of opioid analgesics that are mediated through their peripheral receptors without reversing their analgesic effects. Alvimopan has proven effective in lowering the duration of persistent ileus following abdominal surgery in patients receiving opioid analgesics for postoperative pain control. Methylnaltrexone has proven effective for relief of opioid-induced constipation in patients taking opioid analgesics on a chronic basis.
Opioid and COX Inhibitor Combinations When used in combination, opioids and COX inhibitors have additive effects. Because a lower dose of each can be used to achieve the same degree of pain relief and their side effects are nonadditive, such combinations are used to lower the severity of dose-related side effects. However, fixed-ratio combinations of an opioid with acetaminophen carry an important risk. Dose escalation as a result of increased severity of pain or decreased opioid effect as a result of tolerance may lead to ingestion of levels of acetaminophen that are toxic to the liver. Although acetaminophen-related hepatotoxicity is uncommon, it remains a significant cause for liver failure. Thus, many practitioners have moved away from the use of opioid-acetaminophen combination analgesics to avoid the risk of excessive acetaminophen exposure as the dose of the analgesic is escalated.
CHRONIC PAIN
Managing patients with chronic pain is intellectually and emotionally challenging. The patient’s problem is often difficult or impossible to diagnose with certainty; such patients are demanding of the physician’s time and often appear emotionally distraught. The traditional medical approach of seeking an obscure organic pathology is usually unhelpful. On the other hand, psychological evaluation and behaviorally based treatment paradigms are frequently helpful, particularly in the setting of a multidisciplinary pain-management center. Unfortunately, this approach, while effective, remains largely underused in current medical practice.
There are several factors that can cause, perpetuate, or exacerbate chronic pain. First, of course, the patient may simply have a disease that is characteristically painful for which there is presently no cure. Arthritis, cancer, chronic daily headaches, fibromyalgia, and diabetic neuropathy are examples of this. Second, there may be secondary perpetuating factors that are initiated by disease and persist after that disease has resolved. Examples include damaged sensory nerves, sympathetic efferent activity, and painful reflex muscle contraction (spasm). Finally, a variety of psychological conditions can exacerbate or even cause pain.
There are certain areas to which special attention should be paid in a patient’s medical history. Because depression is the most common emotional disturbance in patients with chronic pain, patients should be questioned about their mood, appetite, sleep patterns, and daily activity. A simple standardized questionnaire, such as the Beck Depression Inventory, can be a useful screening device. It is important to remember that major depression is a common, treatable, and potentially fatal illness.
Other clues that a significant emotional disturbance is contributing to a patient’s chronic pain complaint include pain that occurs in multiple, unrelated sites; a pattern of recurrent, but separate, pain problems beginning in childhood or adolescence; pain beginning at a time of emotional trauma, such as the loss of a parent or spouse; a history of physical or sexual abuse; and past or present substance abuse.
On examination, special attention should be paid to whether the patient guards the painful area and whether certain movements or postures are avoided because of pain. Discovering a mechanical component to the pain can be useful both diagnostically and therapeutically. Painful areas should be examined for deep tenderness, noting whether this is localized to muscle, ligamentous structures, or joints. Chronic myofascial pain is very common, and, in these patients, deep palpation may reveal highly localized trigger points that are firm bands or knots in muscle. Relief of the pain following injection of local anesthetic into these trigger points supports the diagnosis. A neuropathic component to the pain is indicated by evidence of nerve damage, such as sensory impairment, exquisitely sensitive skin (allodynia), weakness, and muscle atrophy, or loss of deep tendon reflexes. Evidence suggesting sympathetic nervous system involvement includes the presence of diffuse swelling, changes in skin color and temperature, and hypersensitive skin and joint tenderness compared with the normal side. Relief of the pain with a sympathetic block supports the diagnosis, but once the condition becomes chronic, the response to sympathetic blockade is of variable magnitude and duration; the role for repeated sympathetic blocks in the overall management of CRPS is not established.
A guiding principle in evaluating patients with chronic pain is to assess both emotional and organic factors before initiating therapy. Addressing these issues together, rather than waiting to address emotional issues after organic causes of pain have been ruled out, improves compliance in part because it assures patients that a psychological evaluation does not mean that the physician is questioning the validity of their complaint. Even when an organic cause for a patient’s pain can be found, it is still wise to look for other factors. For example, a cancer patient with painful bony metastases may have additional pain due to nerve damage and may also be depressed. Optimal therapy requires that each of these factors be looked for and treated.
19 | Chest Discomfort |
Chest discomfort is among the most common reasons for which patients present for medical attention at either an emergency department (ED) or an outpatient clinic. The evaluation of nontraumatic chest discomfort is inherently challenging owing to the broad variety of possible causes, a minority of which are life-threatening conditions that should not be missed. It is helpful to frame the initial diagnostic assessment and triage of patients with acute chest discomfort around three categories: (1) myocardial ischemia; (2) other cardiopulmonary causes (pericardial disease, aortic emergencies, and pulmonary conditions); and (3) non-cardiopulmonary causes. Although rapid identification of high-risk conditions is a priority of the initial assessment, strategies that incorporate routine liberal use of testing carry the potential for adverse effects of unnecessary investigations.
EPIDEMIOLOGY AND NATURAL HISTORY
Chest discomfort is the third most common reason for visits to the ED in the United States, resulting in 6 to 7 million emergency visits each year. More than 60% of patients with this presentation are hospitalized for further testing, and the rest undergo additional investigation in the ED. Fewer than 25% of evaluated patients are eventually diagnosed with acute coronary syndrome (ACS), with rates of 5–15% in most series of unselected populations. In the remainder, the most common diagnoses are gastrointestinal causes (Fig. 19-1), and fewer than 10% are other life-threatening cardiopulmonary conditions. In a large proportion of patients with transient acute chest discomfort, ACS or another acute cardiopulmonary cause is excluded but the cause is not determined. Therefore, the resources and time devoted to the evaluation of chest discomfort in the absence of a severe cause are substantial. Nevertheless, a disconcerting 2–6% of patients with chest discomfort of presumed non-ischemic etiology who are discharged from the ED are later deemed to have had a missed myocardial infarction (MI). Patients with a missed diagnosis of MI have a 30-day risk of death that is double that of their counterparts who are hospitalized.
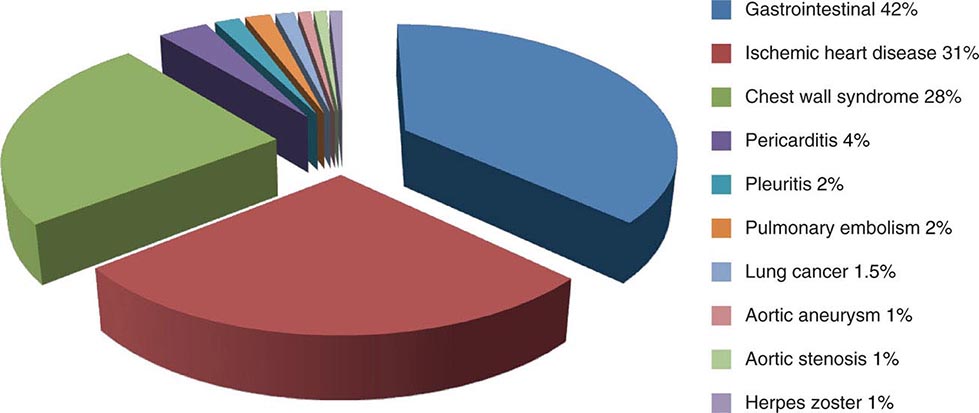
FIGURE 19-1 Distribution of final discharge diagnoses in patients with nontraumatic acute chest pain. (Figure prepared from data in P Fruergaard et al: Eur Heart J 17:1028, 1996.)
The natural histories of ACS, acute pericardial diseases, pulmonary embolism, and aortic emergencies are discussed in Chaps. 288, 294 and 295, 300, and 301, respectively. In a study of more than 350,000 patients with unspecified presumed non-cardiopulmonary chest discomfort, the mortality rate 1 year after discharge was <2% and did not differ significantly from age-adjusted mortality in the general population. The estimated rate of major cardiovascular events through 30 days in patients with acute chest pain who had been stratified as low risk was 2.5% in a large population-based study that excluded patients with ST-segment elevation or definite noncardiac chest pain.
CAUSES OF CHEST DISCOMFORT
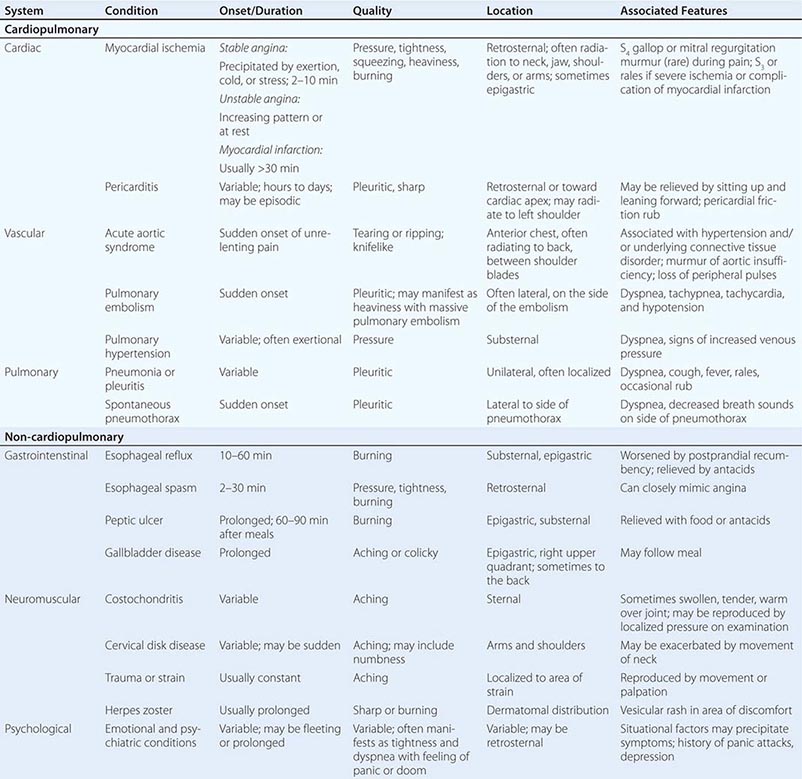
The major etiologies of chest discomfort are discussed in this section and summarized in Table 19-1. Additional elements of the history, physical examination, and diagnostic testing that aid in distinguishing these causes are discussed in a later section (see “Approach to the Patient”).
TYPICAL CLINICAL FEATURES OF MAJOR CAUSES OF ACUTE CHEST DISCOMFORT |
MYOCARDIAL ISCHEMIA/INJURY
Myocardial ischemia causing chest discomfort, termed angina pectoris, is a primary clinical concern in patients presenting with chest symptoms. Myocardial ischemia is precipitated by an imbalance between myocardial oxygen requirements and myocardial oxygen supply, resulting in insufficient delivery of oxygen to meet the heart’s metabolic demands. Myocardial oxygen consumption may be elevated by increases in heart rate, ventricular wall stress, and myocardial contractility, whereas myocardial oxygen supply is determined by coronary blood flow and coronary arterial oxygen content. When myocardial ischemia is sufficiently severe and prolonged in duration (as little as 20 min), irreversible cellular injury occurs, resulting in MI.
Ischemic heart disease is most commonly caused by atheromatous plaque that obstructs one or more of the epicardial coronary arteries. Stable ischemic heart disease (Chap. 293) usually results from the gradual atherosclerotic narrowing of the coronary arteries. Stable angina is characterized by ischemic episodes that are typically precipitated by a superimposed increase in oxygen demand during physical exertion and relieved upon resting. Ischemic heart disease becomes unstable most commonly when rupture or erosion of one or more atherosclerotic lesions triggers coronary thrombosis (Chap. 291e). Unstable ischemic heart disease is classified clinically by the presence or absence of detectable myocardial injury and the presence or absence of ST-segment elevation on the patient’s electrocardiogram (ECG). When acute coronary atherothrombosis occurs, the intracoronary thrombus may be partially obstructive, generally leading to myocardial ischemia in the absence of ST-segment elevation. Marked by ischemic symptoms at rest, with minimal activity, or in an accelerating pattern, unstable ischemic heart disease is classified as unstable angina when there is no detectable myocardial injury and as non–ST elevation MI (NSTEMI) when there is evidence of myocardial necrosis (Chap. 294). When the coronary thrombus is acutely and completely occlusive, transmural myocardial ischemia usually ensues, with ST-segment elevation on the ECG and myocardial necrosis leading to a diagnosis of ST elevation MI (STEMI, see Chap. 295).
Clinicians should be aware that unstable ischemic symptoms may also occur predominantly because of increased myocardial oxygen demand (e.g., during intense psychological stress or fever) or because of decreased oxygen delivery due to anemia, hypoxia, or hypotension. However, the term acute coronary syndrome, which encompasses unstable angina, NSTEMI, and STEMI, is in general reserved for ischemia precipitated by acute coronary atherothrombosis. In order to guide therapeutic strategies, a standardized system for classification of MI has been expanded to discriminate MI resulting from acute coronary thrombosis (type 1) from MI occurring secondary to other imbalances of myocardial oxygen supply and demand (type 2; see Chap. 294).
Other contributors to stable and unstable ischemic heart disease, such as endothelial dysfunction, microvascular disease, and vasospasm, may exist alone or in combination with coronary atherosclerosis and may be the dominant cause of myocardial ischemia in some patients. Moreover, non-atherosclerotic processes, including congenital abnormalities of the coronary vessels, myocardial bridging, coronary arteritis, and radiation-induced coronary disease, can lead to coronary obstruction. In addition, conditions associated with extreme myocardial oxygen demand and impaired endocardial blood flow, such as aortic valve disease (Chap. 301), hypertrophic cardiomyopathy, or idiopathic dilated cardiomyopathy (Chap. 287), can precipitate myocardial ischemia in patients with or without underlying obstructive atherosclerosis.
Characteristics of Ischemic Chest Discomfort The clinical characteristics of angina pectoris, often referred to simply as “angina,” are highly similar whether the ischemic discomfort is a manifestation of stable ischemic heart disease, unstable angina, or MI; the exceptions are differences in the pattern and duration of symptoms associated with these syndromes (Table 19-1). Heberden initially described angina as a sense of “strangling and anxiety.” Chest discomfort characteristic of myocardial ischemia is typically described as aching, heavy, squeezing, crushing, or constricting. However, in a substantial minority of patients, the quality of discomfort is extremely vague and may be described as a mild tightness, or merely an uncomfortable feeling, that sometimes is experienced as numbness or a burning sensation. The site of the discomfort is usually retrosternal, but radiation is common and generally occurs down the ulnar surface of the left arm; the right arm, both arms, neck, jaw, or shoulders may also be involved. These and other characteristics of ischemic chest discomfort pertinent to discrimination from other causes of chest pain are discussed later in this chapter (see “Approach to the Patient”).
Stable angina usually begins gradually and reaches its maximal intensity over a period of minutes before dissipating within several minutes with rest or with nitroglycerin. The discomfort typically occurs predictably at a characteristic level of exertion or psychological stress. By definition, unstable angina is manifest by self-limited anginal chest discomfort that is exertional but occurs at increased frequency with progressively lower intensity of physical activity or even at rest. Chest discomfort associated with MI is typically more severe, is prolonged (usually lasting ≥30 min), and is not relieved by rest.
Mechanisms of Cardiac Pain The neural pathways involved in ischemic cardiac pain are poorly understood. Ischemic episodes are thought to excite local chemosensitive and mechanoreceptive receptors that, in turn, stimulate release of adenosine, bradykinin, and other substances that activate the sensory ends of sympathetic and vagal afferent fibers. The afferent fibers traverse the nerves that connect to the upper five thoracic sympathetic ganglia and upper five distal thoracic roots of the spinal cord. From there, impulses are transmitted to the thalamus. Within the spinal cord, cardiac sympathetic afferent impulses may converge with impulses from somatic thoracic structures, and this convergence may be the basis for referred cardiac pain. In addition, cardiac vagal afferent fibers synapse in the nucleus tractus solitarius of the medulla and then descend to the upper cervical spinothalamic tract, and this route may contribute to anginal pain experienced in the neck and jaw.
OTHER CARDIOPULMONARY CAUSES
Pericardial and Other Myocardial Diseases (See also Chap. 288) Inflammation of the pericardium due to infectious or noninfectious causes can be responsible for acute or chronic chest discomfort. The visceral surface and most of the parietal surface of the pericardium are insensitive to pain. Therefore, the pain of pericarditis is thought to arise principally from associated pleural inflammation and is more common with infectious causes of pericarditis, which typically involve the pleura. Because of this pleural association, the discomfort of pericarditis is usually pleuritic pain that is exacerbated by breathing, coughing, or changes in position. Moreover, owing to the overlapping sensory supply of the central diaphragm via the phrenic nerve with somatic sensory fibers originating in the third to fifth cervical segments, the pain of pleural pericarditis is often referred to the shoulder and neck. Involvement of the pleural surface of the lateral diaphragm can lead to pain in the upper abdomen.
Acute inflammatory and other non-ischemic myocardial diseases can also produce chest discomfort. The symptoms of Takotsubo (stress-related) cardiomyopathy often start abruptly with chest pain and shortness of breath. This form of cardiomyopathy, in its most recognizable form, is triggered by an emotionally or physically stressful event and may mimic acute MI because of its commonly associated ECG abnormalities, including ST-segment elevation, and elevated biomarkers of myocardial injury. Observational studies support a predilection for women >50 years of age. The symptoms of acute myocarditis are highly varied. Chest discomfort may either originate with inflammatory injury of the myocardium or be due to severe increases in wall stress related to poor ventricular performance.
Diseases of the Aorta (See also Chap. 301) Acute aortic dissection (Fig. 19-1) is a less common cause of chest discomfort but is important because of the catastrophic natural history of certain subsets of cases when recognized late or left untreated. Acute aortic syndromes encompass a spectrum of acute aortic diseases related to disruption of the media of the aortic wall. Aortic dissection involves a tear in the aortic intima, resulting in separation of the media and creation of a separate “false” lumen. A penetrating ulcer has been described as ulceration of an aortic atheromatous plaque that extends through the intima and into the aortic media, with the potential to initiate an intramedial dissection or rupture into the adventitia. Intramural hematoma is an aortic wall hematoma with no demonstrable intimal flap, no radiologically apparent intimal tear, and no false lumen. Intramural hematoma can occur due to either rupture of the vasa vasorum or, less commonly, a penetrating ulcer.
Each of these subtypes of acute aortic syndrome typically presents with chest discomfort that is often severe, sudden in onset, and sometimes described as “tearing” in quality. Acute aortic syndromes involving the ascending aorta tend to cause pain in the midline of the anterior chest, whereas descending aortic syndromes most often present with pain in the back. Therefore, dissections that begin in the ascending aorta and extend to the descending aorta tend to cause pain in the front of the chest that extends toward the back, between the shoulder blades. Proximal aortic dissections that involve the ascending aorta (type A in the Stanford nomenclature) are at high risk for major complications that may influence the clinical presentation, including (1) compromise of the aortic ostia of the coronary arteries, resulting in MI; (2) disruption of the aortic valve, causing acute aortic insufficiency; and (3) rupture of the hematoma into the pericardial space, leading to pericardial tamponade.
Knowledge of the epidemiology of acute aortic syndromes can be helpful in maintaining awareness of this relatively uncommon group of disorders (estimated annual incidence, 3 cases per 100,000 population). Nontraumatic aortic dissections are very rare in the absence of hypertension or conditions associated with deterioration of the elastic or muscular components of the aortic media, including pregnancy, bicuspid aortic disease, or inherited connective tissue diseases, such as Marfan and Ehlers-Danlos syndromes.
Although aortic aneurysms are most often asymptomatic, thoracic aortic aneurysms can cause chest pain and other symptoms by compressing adjacent structures. This pain tends to be steady, deep, and occasionally severe. Aortitis, whether of noninfectious or infectious etiology, in the absence of aortic dissection is a rare cause of chest or back discomfort.
Pulmonary Conditions Pulmonary and pulmonary-vascular conditions that cause chest discomfort usually do so in conjunction with dyspnea and often produce symptoms that have a pleuritic nature.
PULMONARY EMBOLISM (See also Chap. 300) Pulmonary emboli (annual incidence, ~1 per 1000) can produce dyspnea and chest discomfort that is sudden in onset. Typically pleuritic in pattern, the chest discomfort associated with pulmonary embolism may result from (1) involvement of the pleural surface of the lung adjacent to a resultant pulmonary infarction; (2) distention of the pulmonary artery; or (3) possibly, right ventricular wall stress and/or subendocardial ischemia related to acute pulmonary hypertension. The pain associated with small pulmonary emboli is often lateral and pleuritic and is believed to be related to the first of these three possible mechanisms. In contrast, massive pulmonary emboli may cause severe substernal pain that may mimic an MI and that is plausibly attributed to the second and third of these potential mechanisms. Massive or submassive pulmonary embolism may also be associated with syncope, hypotension, and signs of right heart failure. Other typical characteristics that aid in the recognition of pulmonary embolism are discussed later in this chapter (see “Approach to the Patient”).
PNEUMOTHORAX (See also Chap. 317) Primary spontaneous pneumothorax is a rare cause of chest discomfort, with an estimated annual incidence in the United States of 7 per 100,000 among men and <2 per 100,000 among women. Risk factors include male sex, smoking, family history, and Marfan syndrome. The symptoms are usually sudden in onset, and dyspnea may be mild; thus, presentation to medical attention is sometimes delayed. Secondary spontaneous pneumothorax may occur in patients with underlying lung disorders, such as chronic obstructive pulmonary disease, asthma, or cystic fibrosis, and usually produces symptoms that are more severe. Tension pneumothorax is a medical emergency caused by trapped intrathoracic air that precipitates hemodynamic collapse.
Other Pulmonary Parenchymal, Pleural, or Vascular Disease (See also Chaps. 304, 305, and 316) Most pulmonary diseases that produce chest pain, including pneumonia and malignancy, do so because of involvement of the pleura or surrounding structures. Pleurisy is typically described as a knifelike pain that is worsened by inspiration or coughing. In contrast, chronic pulmonary hypertension can manifest as chest pain that may be very similar to angina in its characteristics, suggesting right ventricular myocardial ischemia in some cases. Reactive airways diseases similarly can cause chest tightness associated with breathlessness rather than pleurisy.
NON-CARDIOPULMONARY CAUSES
Gastrointenstinal Conditions (See also Chap. 344) Gastrointestinal disorders are the most common cause of nontraumatic chest discomfort and often produce symptoms that are difficult to discern from more serious causes of chest pain, including myocardial ischemia. Esophageal disorders, in particular, may simulate angina in the character and location of the pain. Gastroesophageal reflux and disorders of esophageal motility are common and should be considered in the differential diagnosis of chest pain (Fig. 19-1 and Table 19-1). Acid reflux often causes a burning discomfort. The pain of esophageal spasm, in contrast, is commonly an intense, squeezing discomfort that is retrosternal in location and, like angina, may be relieved by nitroglycerin or dihydropyridine calcium channel antagonists. Chest pain can also result from injury to the esophagus, such as a Mallory-Weiss tear or even an esophageal rupture (Boerhaave syndrome) caused by severe vomiting. Peptic ulcer disease is most commonly epigastric in location but can radiate into the chest (Table 19-1).
Hepatobiliary disorders, including cholecystitis and biliary colic, may mimic acute cardiopulmonary diseases. Although the pain arising from these disorders usually localizes to the right upper quadrant of the abdomen, it is variable and may be felt in the epigastrium and radiate to the back and lower chest. This discomfort is sometimes referred to the scapula or may in rare cases be felt in the shoulder, suggesting diaphragmatic irritation. The pain is steady, usually lasts several hours, and subsides spontaneously, without symptoms between attacks. Pain resulting from pancreatitis is typically aching epigastric pain that radiates to the back.
Musculoskeletal and Other Causes (See also Chap. 393) Chest discomfort can be produced by any musculoskeletal disorder involving the chest wall or the nerves of the chest wall, neck, or upper limbs. Costochondritis causing tenderness of the costochondral junctions (Tietze’s syndrome) is relatively common. Cervical radiculitis may manifest as a prolonged or constant aching discomfort in the upper chest and limbs. The pain may be exacerbated by motion of the neck. Occasionally, chest pain can be caused by compression of the brachial plexus by the cervical ribs, and tendinitis or bursitis involving the left shoulder may mimic the radiation of angina. Pain in a dermatomal distribution can also be caused by cramping of intercostal muscles or by herpes zoster (Chap. 217).
Emotional and Psychiatric Conditions As many as 10% of patients who present to emergency departments with acute chest discomfort have a panic disorder or related condition (Table 19-1). The symptoms may include chest tightness or aching that is associated with a sense of anxiety and difficulty breathing. The symptoms may be prolonged or fleeting.
CONSIDERATIONS IN THE ASSESSMENT OF THE PATIENT WITH CHEST DISCOMFORT |

The evaluation of nontraumatic chest discomfort relies heavily on the clinical history and physical examination to direct subsequent diagnostic testing. The evaluating clinician should assess the quality, location (including radiation), and pattern (including onset and duration) of the pain as well as any provoking or alleviating factors. The presence of associated symptoms may also be useful in establishing a diagnosis.
Quality of Pain The quality of chest discomfort alone is never sufficient to establish a diagnosis. However, the characteristics of the pain are pivotal in formulating an initial clinical impression and assessing the likelihood of a serious cardiopulmonary process (Table 19-1), including acs in particular (Fig. 19-2). Pressure or tightness is consistent with a typical presentation of myocardial ischemic pain. Nevertheless, the clinician must remember that some patients with ischemic chest symptoms deny any “pain” but rather complain of dyspnea or a vague sense of anxiety. The severity of the discomfort has poor diagnostic accuracy. It is often helpful to ask about the similarity of the discomfort to previous definite ischemic symptoms. It is unusual for angina to be sharp, as in knifelike, stabbing, or pleuritic; however, patients sometimes use the word “sharp” to convey the intensity of discomfort rather than the quality. Pleuritic discomfort is suggestive of a process involving the pleura, including pericarditis, pulmonary embolism, or pulmonary parenchymal processes. Less frequently, the pain of pericarditis or massive pulmonary embolism is a steady severe pressure or aching that can be difficult to discriminate from myocardial ischemia. “Tearing” or “ripping” pain is often described by patients with acute aortic dissection. However, acute aortic emergencies also present commonly with severe, knifelike pain. A burning quality can suggest acid reflux or peptic ulcer disease but may also occur with myocardial ischemia. Esophageal pain, particularly with spasm, can be a severe squeezing discomfort identical to angina.
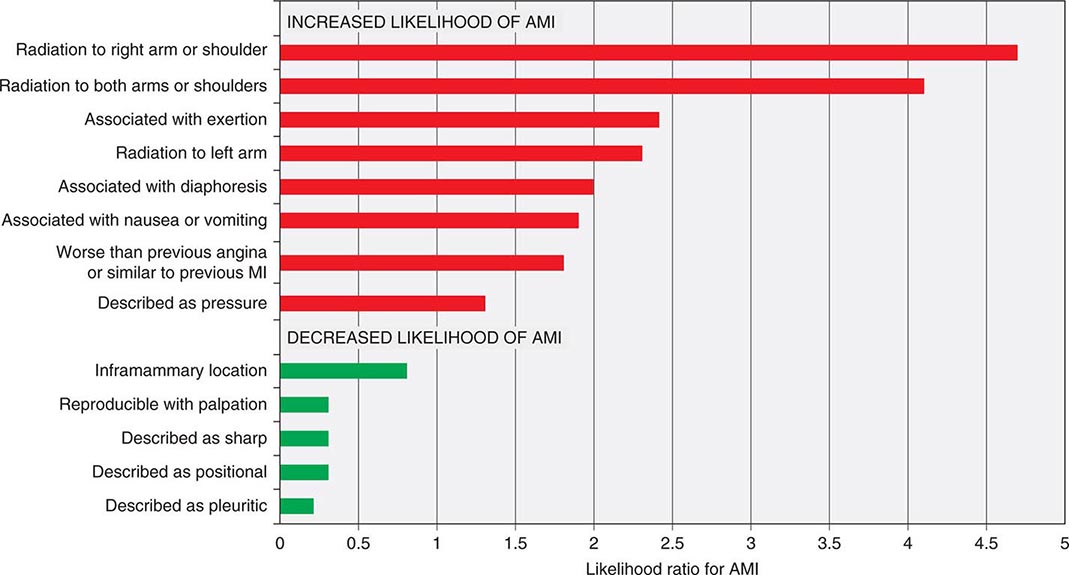
FIGURE 19-2 Association of chest pain characteristics with the probability of acute myocardial infarction (AMI). (Figure prepared from data in CJ Swap, JT Nagurney: JAMA 294:2623, 2005.)
Location of Discomfort A substernal location with radiation to the neck, jaw, shoulder, or arms is typical of myocardial ischemic discomfort. Some patients present with aching in sites of radiated pain
as their only symptoms of ischemia. However, pain that is highly localized—e.g., that which can be demarcated by the tip of one finger—is highly unusual for angina. A retrosternal location should prompt consideration of esophageal pain; however, other gastrointestinal conditions usually present with pain that is most intense in the abdomen or epigastrium, with possible radiation into the chest. Angina may also occur in an epigastric location. However, pain that occurs solely above the mandible or below the epigastrium is rarely angina. Severe pain radiating to the back, particularly between the shoulder blades, should prompt consideration of an acute aortic syndrome. Radiation to the trapezius ridge is characteristic of pericardial pain and does not usually occur with angina.
Pattern Myocardial ischemic discomfort usually builds over minutes and is exacerbated by activity and mitigated by rest. In contrast, pain that reaches its peak intensity immediately is more suggestive of aortic dissection, pulmonary embolism, or spontaneous pneumothorax. Pain that is fleeting (lasting only a few seconds) is rarely ischemic in origin. Similarly, pain that is constant in intensity for a prolonged period (many hours to days) is unlikely to represent myocardial ischemia if it occurs in the absence of other clinical consequences, such as abnormalities of the ECG, elevation of cardiac biomarkers, or clinical sequelae (e.g., heart failure or hypotension). Both myocardial ischemia and acid reflux may have their onset in the morning, the latter because of the absence of food to absorb gastric acid.
Provoking and Alleviating Factors Patients with myocardial ischemic pain usually prefer to rest, sit, or stop walking. However, clinicians should be aware of the phenomenon of “warm-up angina” in which some patients experience relief of angina as they continue at the same or even a greater level of exertion without symptoms (Chap. 293). Alterations in the intensity of pain with changes in position or movement of the upper extremities and neck are less likely with myocardial ischemia and suggest a musculoskeletal etiology. The pain of pericarditis, however, often is worse in the supine position and relieved by sitting upright and leaning forward. Gastroesophageal reflux may be exacerbated by alcohol, some foods, or by a reclined position. Relief can occur with sitting.
Exacerbation by eating suggests a gastrointestinal etiology such as peptic ulcer disease, cholecystitis, or pancreatitis. Peptic ulcer disease tends to become symptomatic 60–90 min after meals. However, in the setting of severe coronary atherosclerosis, redistribution of blood flow to the splanchnic vasculature after eating can trigger postprandial angina. The discomfort of acid reflux and peptic ulcer disease is usually diminished promptly by acid-reducing therapies. In contrast with its impact in some patients with angina, physical exertion is very unlikely to alter symptoms from gastrointestinal causes of chest pain. Relief of chest discomfort within minutes after administration of nitroglycerin is suggestive of but not sufficiently sensitive or specific for a definitive diagnosis of myocardial ischemia. Esophageal spasm may also be relieved promptly with nitroglycerin. A delay of >10 min before relief is obtained after nitroglycerin suggests that the symptoms either are not caused by ischemia or are caused by severe ischemia, such as during acute MI.
Associated Symptoms Symptoms that accompany myocardial ischemia may include diaphoresis, dyspnea, nausea, fatigue, faintness, and eructations. In addition, these symptoms may exist in isolation as anginal equivalents (i.e., symptoms of myocardial ischemia other than typical angina), particularly in women and the elderly. Dyspnea may occur with multiple conditions considered in the differential diagnosis of chest pain and thus is not discriminative, but the presence of dyspnea is important because it suggests a cardiopulmonary etiology. Sudden onset of significant respiratory distress should lead to consideration of pulmonary embolism and spontaneous pneumothorax. Hemoptysis may occur with pulmonary embolism, or as blood-tinged frothy sputum in severe heart failure but usually points toward a pulmonary parenchymal etiology of chest symptoms. Presentation with syncope or pre-syncope should prompt consideration of hemodynamically significant pulmonary embolism or aortic dissection as well as ischemic arrhythmias. Although nausea and vomiting suggest a gastrointestinal disorder, these symptoms may occur in the setting of MI (more commonly inferior MI), presumably because of activation of the vagal reflex or stimulation of left ventricular receptors as part of the Bezold-Jarisch reflex.
Past Medical History The past medical history is useful in assessing the patient for risk factors for coronary atherosclerosis (Chap. 291e) and venous thromboembolism (Chap. 300) as well as for conditions that may predispose the patient to specific disorders. For example, a history of connective tissue diseases such as marfan syndrome should heighten the clinician’s suspicion of an acute aortic syndrome or spontaneous pneumothorax. A careful history may elicit clues about depression or prior panic attacks.
PHYSICAL EXAMINATION
In addition to providing an initial assessment of the patient’s clinical stability, the physical examination of patients with chest discomfort can provide direct evidence of specific etiologies of chest pain (e.g., unilateral absence of lung sounds) and can identify potential precipitants of acute cardiopulmonary causes of chest pain (e.g., uncontrolled hypertension), relevant comorbid conditions (e.g., obstructive pulmonary disease), and complications of the presenting syndrome (e.g., heart failure). However, because the findings on physical examination may be normal in patients with unstable ischemic heart disease, an unremarkable physical exam is not definitively reassuring.
General The patient’s general appearance is helpful in establishing an initial impression of the severity of illness. Patients with acute MI or other acute cardiopulmonary disorders often appear anxious, uncomfortable, pale, cyanotic, or diaphoretic. Patients who are massaging or clutching their chests may describe their pain with a clenched fist held against the sternum (Levine’s sign). Occasionally, body habitus is helpful—e.g., in patients with Marfan syndrome or the prototypical young, tall, thin man with spontaneous pneumothorax.
Vital Signs Significant tachycardia and hypotension are indicative of important hemodynamic consequences of the underlying cause of chest discomfort and should prompt a rapid survey for the most severe conditions, such as acute MI with cardiogenic shock, massive pulmonary embolism, pericarditis with tamponade, or tension pneumothorax. Acute aortic emergencies usually present with severe hypertension but may be associated with profound hypotension when there is coronary arterial compromise or dissection into the pericardium. Sinus tachycardia is an important manifestation of submassive pulmonary embolism. Tachypnea and hypoxemia point toward a pulmonary cause. The presence of low-grade fever is nonspecific because it may occur with MI and with thromboembolism in addition to infection.
Pulmonary Examination of the lungs may localize a primary pulmonary cause of chest discomfort, as in cases of pneumonia, asthma, or pneumothorax. Left ventricular dysfunction from severe ischemia/infarction as well as acute valvular complications of MI or aortic dissection can lead to pulmonary edema, which is an indicator of high risk.
Cardiac The jugular venous pulse is often normal in patients with acute myocardial ischemia but may reveal characteristic patterns with pericardial tamponade or acute right ventricular dysfunction (Chaps. 267 and 288). Cardiac auscultation may reveal a third or, more commonly, a fourth heart sound, reflecting myocardial systolic or diastolic dysfunction. Murmurs of mitral regurgitation or a harsh murmur of a ventricular-septal defect may indicate mechanical complications of STEMI. A murmur of aortic insufficiency may be a complication of proximal aortic dissection. Other murmurs may reveal underlying cardiac disorders contributory to ischemia (e.g., aortic stenosis or hypertrophic cardiomyopathy). Pericardial friction rubs reflect pericardial inflammation.
Abdominal Localizing tenderness on the abdominal exam is useful in identifying a gastrointestinal cause of the presenting syndrome. Abdominal findings are infrequent with purely acute cardiopulmonary problems, except in the case of underlying chronic cardiopulmonary disease or severe right ventricular dysfunction leading to hepatic congestion.
Vascular Pulse deficits may reflect underlying chronic atherosclerosis, which increases the likelihood of coronary artery disease. However, evidence of acute limb ischemia with loss of the pulse and pallor, particularly in the upper extremities, can indicate catastrophic consequences of aortic dissection. Unilateral lower-extremity swelling should raise suspicion about venous thromboembolism.
Musculoskeletal Pain arising from the costochondral and chondrosternal articulations may be associated with localized swelling, redness, or marked localized tenderness. Pain on palpation of these joints is usually well localized and is a useful clinical sign, though deep palpation may elicit pain in the absence of costochondritis. Although palpation of the chest wall often elicits pain in patients with various musculoskeletal conditions, it should be appreciated that chest wall tenderness does not exclude myocardial ischemia. Sensory deficits in the upper extremities may be indicative of cervical disk disease.
ELECTROCARDIOGRAPHY
Electrocardiography is crucial in the evaluation of nontraumatic chest discomfort. The ECG is pivotal for identifying patients with ongoing ischemia as the principal reason for their presentation as well as secondary cardiac complications of other disorders. Professional society guidelines recommend that an ECG be obtained within 10 min of presentation, with the primary goal of identifying patients with ST-segment elevation diagnostic of MI who are candidates for immediate interventions to restore flow in the occluded coronary artery. ST-segment depression and symmetric T-wave inversions at least 0.2 mV in depth are useful for detecting myocardial ischemia in the absence of STEMI and are also indicative of higher risk of death or recurrent ischemia. Serial performance of ECGs (every 30–60 min) is recommended in the ED evaluation of suspected ACS. In addition, an ECG with right-sided lead placement should be considered in patients with clinically suspected ischemia and a nondiagnostic standard 12-lead ECG. Despite the value of the resting ECG, its sensitivity for ischemia is poor—as low as 20% in some studies.
Abnormalities of the ST segment and T wave may occur in a variety of conditions, including pulmonary embolism, ventricular hypertrophy, acute and chronic pericarditis, myocarditis, electrolyte imbalance, and metabolic disorders. Notably, hyperventilation associated with panic disorder can also lead to nonspecific ST and T-wave abnormalities. Pulmonary embolism is most often associated with sinus tachycardia but can also lead to rightward shift of the ECG axis, manifesting as an S-wave in lead I, with a Q-wave and T-wave in lead III (Chaps. 268 and 300). In patients with ST-segment elevation, the presence of diffuse lead involvement not corresponding to a specific coronary anatomic distribution and PR-segment depression can aid in distinguishing pericarditis from acute MI.
CHEST RADIOGRAPHY
(See Chap. 308e) Plain radiography of the chest is performed routinely when patients present with acute chest discomfort and selectively when individuals who are being evaluated as outpatients have subacute or chronic pain. The chest radiograph is most useful for identifying pulmonary processes, such as pneumonia or pneumothorax. Findings are often unremarkable in patients with ACS, but pulmonary edema may be evident. Other specific findings include widening of the mediastinum in some patients with aortic dissection, Hampton’s hump or Westermark’s sign in patients with pulmonary embolism (Chaps. 300 and 308e), or pericardial calcification in chronic pericarditis.
CARDIAC BIOMARKERS
Laboratory testing in patients with acute chest pain is focused on the detection of myocardial injury. Such injury can be detected by the presence of circulating proteins released from damaged myocardial cells. Owing to the time necessary for this release, initial biomarkers of injury may be in the normal range, even in patients with STEMI. Because of superior cardiac tissue-specificity compared with creatine kinase MB, cardiac troponin is the preferred biomarker for the diagnosis of MI and should be measured in all patients with suspected ACS at presentation and repeated in 3–6 h. Testing after 6 h is required only when there is uncertainty regarding the onset of pain or when stuttering symptoms have occurred. It is not necessary or advisable to measure troponin in patients without suspicion of ACS unless this test is being used specifically for risk stratification (e.g., in pulmonary embolism or heart failure).
The development of cardiac troponin assays with progressively greater analytical sensitivity has facilitated detection of substantially lower blood concentrations of troponin than was previously possible. This evolution permits earlier detection of myocardial injury, enhances the overall accuracy of a diagnosis of MI, and improves risk stratification in suspected ACS. The greater negative predictive value of a negative troponin result with current-generation assays is an advantage in the evaluation of chest pain in the ED. Rapid rule-out protocols that use serial testing and changes in troponin concentration over as short a period as 1–2 h appear promising and remain under investigation. However, with these advantages has come a trade-off: myocardial injury is detected in a larger proportion of patients who have non-ACS cardiopulmonary conditions than with previous, less sensitive assays. This evolution in testing for myocardial necrosis has rendered other aspects of the clinical evaluation critical to the practitioner’s determination of the probability that the symptoms represent ACS. In addition, observation of a change in cardiac troponin concentration between serial samples is useful in discriminating acute causes of myocardial injury from chronic elevation due to underlying structural heart disease, end-stage renal disease, or interfering antibodies. The diagnosis of MI is reserved for acute myocardial injury that is marked by a rising and/or falling pattern—with at least one value exceeding the 99th percentile reference limit—and that is caused by ischemia. Other non-ischemic insults, such as myocarditis, may result in myocardial injury but should not be labeled MI.
Other laboratory assessments may include the D-dimer test to aid in exclusion of pulmonary embolism (Chap. 300). Measurement of a B-type natriuretic peptide is useful when considered in conjunction with the clinical history and exam for the diagnosis of heart failure. B-type natriuretic peptides also provide prognostic information regarding patients with ACS and those with pulmonary embolism. Other putative biomarkers of acute myocardial ischemia or ACS, such as myeloperoxidase, have not been adopted in routine use.
INTEGRATIVE DECISION-AIDS
Multiple clinical algorithms have been developed to aid in decision-making during the evaluation and disposition of patients with acute nontraumatic chest pain. Such decision-aids have been derived on the basis of their capacity to estimate either of two closely related but not identical probabilities: (1) the probability of a final diagnosis of ACS and (2) the probability of major cardiac events during short-term follow-up. Such decision-aids are used most commonly to identify patients with a low clinical probability of ACS who are candidates either for early provocative testing for ischemia or for discharge from the ED. Goldman and Lee developed one of the first such decision-aids, using only the ECG and risk indicators—hypotension, pulmonary rales, and known ischemic heart disease—to categorize patients into four risk categories ranging from a <1% to a >16% probability of a major cardiovascular complication. The Acute Cardiac Ischemia Time-Insensitive Predictive Instrument (ACI-TIPI) combines age, sex, chest pain presence, and ST-segment abnormalities to define a probability of ACS. More recently developed decision-aids are shown in Fig. 19-3. Elements common to each of these tools are (1) symptoms typical for ACS; (2) older age; (3) risk factors for or known atherosclerosis; (4) ischemic ECG abnormalities; and (5) elevated cardiac troponin levels. Although, because of very low specificity, the overall diagnostic performance of such decision-aids is poor (area under the receiver operating curve, 0.55–0.65), they can help identify patients with a very low probability of ACS (e.g., <1%). Nevertheless, no such decision-aid (or single clinical factor) is sufficiently sensitive and well validated to use as a sole tool for clinical decision-making.
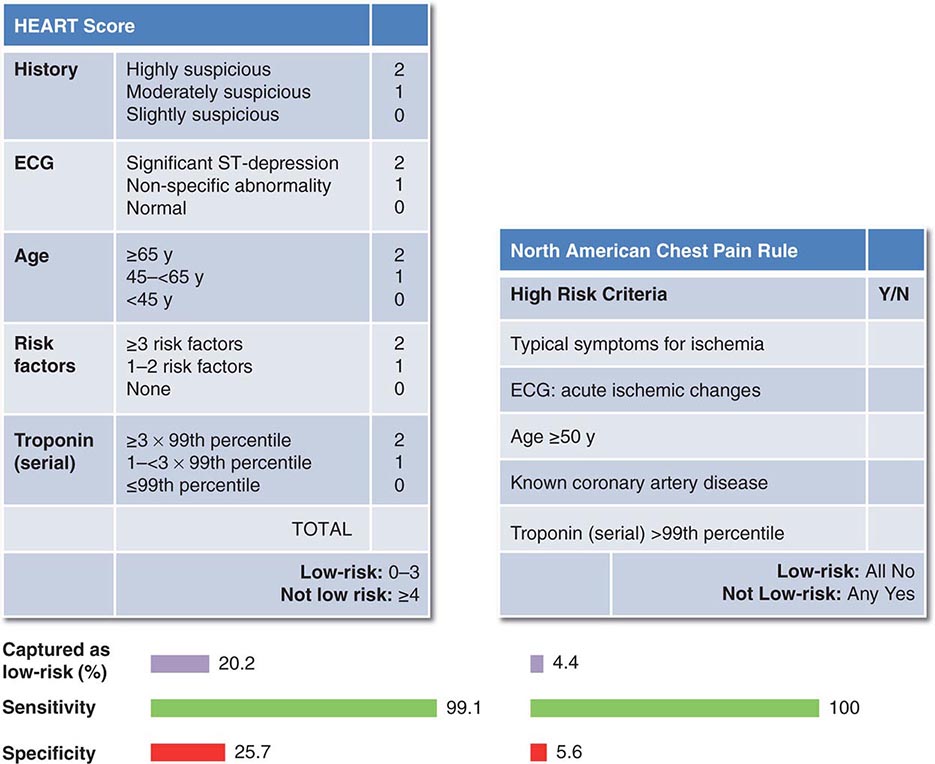
FIGURE 19-3 Examples of decision-aids used in conjunction with serial measurement of cardiac troponin for evaluation of acute chest pain. (Figure prepared from data in SA Mahler et al: Int J Cardiol 168:795, 2013.)
Clinicians should differentiate between the algorithms discussed above and risk scores derived for stratification of prognosis (e.g., the TIMI and GRACE risk scores, Chap. 295) in patients who already have an established diagnosis of ACS. The latter risk scores were not designed to be used for diagnostic assessment.
PROVOCATIVE TESTING FOR ISCHEMIA
Exercise electrocardiography (“stress testing”) is commonly employed for completion of risk stratification of patients who have undergone an initial evaluation that has not revealed a specific cause of chest discomfort and has identified them as being at low or selectively intermediate risk of ACS. Early exercise testing is safe in patients without high-risk findings after 8–12 h of observation and can assist in refining their prognostic assessment. For example, of low-risk patients who underwent exercise testing in the first 48 h after presentation, those without evidence of ischemia had a 2% rate of cardiac events through 6 months, whereas the rate was 15% among patients with either clear evidence of ischemia or an equivocal result. Patients who are unable to exercise may undergo pharmacological stress testing with either nuclear perfusion imaging or echocardiography. Notably, some experts have deemed the routine use of stress testing for low-risk patients unsupported by direct clinical evidence and a potentially unnecessary source of cost.
Professional society guidelines identify ongoing chest pain as a contraindication to stress testing. In selected patients with persistent pain and nondiagnostic ECG and biomarker data, resting myocardial perfusion images can be obtained; the absence of any perfusion abnormality substantially reduces the likelihood of coronary artery disease. In some centers, early myocardial perfusion imaging is performed as part of a routine strategy for evaluating patients at low or intermediate risk of ACS in parallel with other testing. Management of patients with normal perfusion images can be expedited with earlier discharge and outpatient stress testing, if indicated. Those with abnormal rest perfusion imaging, which cannot discriminate between old or new myocardial defects, must undergo additional in-hospital evaluation.
OTHER NONINVASIVE STUDIES
Other noninvasive imaging studies of the chest can be used selectively to provide additional diagnostic and prognostic information on patients with chest discomfort.
Echocardiography Echocardiography is not necessarily routine in patients with chest discomfort. However, in patients with an uncertain diagnosis, particularly those with nondiagnostic ST elevation, ongoing symptoms, or hemodynamic instability, detection of abnormal regional wall motion provides evidence of possible ischemic dysfunction. Echocardiography is diagnostic in patients with mechanical complications of MI or in patients with pericardial tamponade. Transthoracic echocardiography is poorly sensitive for aortic dissection, although an intimal flap may sometimes be detected in the ascending aorta.
CT Angiography (See Chap. 270e) CT angiography is emerging as a modality for the evaluation of patients with acute chest discomfort. Coronary CT angiography is a sensitive technique for detection of obstructive coronary disease, particularly in the proximal third of the major epicardial coronary arteries. CT appears to enhance the speed to disposition of patients with a low-intermediate probability for ACS; its major strength being the negative predictive value of a finding of no significant disease. In addition, contrast-enhanced CT can detect focal areas of myocardial injury in the acute setting as decreased areas of enhancement. At the same time, CT angiography can exclude aortic dissection, pericardial effusion, and pulmonary embolism. Balancing factors in the consideration of the emerging role of coronary CT angiography in low-risk patients are radiation exposure and additional testing prompted by nondiagnostic abnormal results.
MRI (See Chap. 270e) Cardiac magnetic resonance (CMR) imaging is an evolving, versatile technique for structural and functional evaluation of the heart and the vasculature of the chest. CMR accurately measures ventricular dimensions and function and can be performed as a modality for pharmacologic stress perfusion imaging. Gadolinium-enhanced CMR can provide early detection of MI, defining areas of myocardial necrosis accurately, and can delineate patterns of myocardial disease that are often useful in discriminating ischemic from non-ischemic myocardial injury. Although usually not practical for the urgent evaluation of acute chest discomfort, CMR can be a useful modality for cardiac structural evaluation of patients with elevated cardiac troponin levels in the absence of definite coronary artery disease. CMR coronary angiography is in its early stages. MRI also permits highly accurate assessment for aortic dissection but is infrequently used as the first test because CT and transesophageal echocardiography are usually more practical.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


