B-cell neoplasms are a heterogeneous group of clinicopathologic entities, ranging from indolent to highly aggressive (Table 23.1). Those composed predominantly of mature small B-cells may be particularly difficult to distinguish from one another in the bone marrow (Table 23.2).
Certain B-cell neoplasms are discussed with plasma cell neoplasms, as they share many similarities.
Plasmablastic lymphoma is discussed in Chapter 25, as its clinical and pathologic features and differential diagnosis are closely linked with plasma cell neoplasms.
Clonality is not always easy to assess in B-cell neoplasms (1,2). Many cases of dual clonality, and a rare case of triclonality, have been reported. The morphology in such cases may appear to represent one disease, as in biclonal B-cell chronic lymphocytic leukemia, or a combination of diseases, as in B-cell chronic lymphocytic leukemia and multiple myeloma. Conversely, the presence of two different immunoglobulin light chains is not proof of biclonality, as occasional cases of monoclonal B-cell neoplasms have been shown to express both light chains.
Following are entities which may show bone marrow involvement. The categories do not exactly match the WHO 2008 list, and they are listed in alphabetical order rather than WHO order. See Appendix A for WHO 2008 classifications.
PRECURSOR B-CELL NEOPLASM
B-Cell Lymphoblastic Lymphoma
B-cell lymphoblastic lymphoma (B-LBL) usually occurs de novo, but has also been reported as a terminal phase of B-cell chronic lymphocytic leukemia and follicular lymphoma, and as a complication of solid organ transplantation (3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41). It accounts for about 10% of LBL cases, the remainder being of precursor T-cell phenotype (see also Chapter 19).
Patients may present at any age, from infancy to adulthood. Most cases occur in children. B-LBL presents as a tumor which often involves the soft tissues of the head and neck, but may also present as a mass of the skin and subcutaneous tissue, meninges and cerebrospinal fluid, breast, heart, pleura and pleural space, retroperitoneum, kidney, genital tract, and bone. Cases with 20% or more blasts in the peripheral blood or bone marrow at the time of diagnosis are better classified as precursor B-cell acute lymphoblastic leukemia (ALL). Lymphoblastic malignancy presenting as a lytic bone lesion may be interpreted as either B-LBL or precursor B-cell ALL.
Peripheral blood and bone marrow aspirate smears and histologic sections of the bone marrow biopsy show, in the rare involved case, less than 20% blasts (see Figs. 19.5 and 19.12). The morphologic findings are identical to precursor B-cell ALL. The blasts are small to moderate-sized cells with round, sometimes cleaved or convoluted, nuclei and scant cytoplasm. Rosette formation is not typical but has been reported. The cells may be positive for periodic acid-Schiff (PAS).
Immunophenotyping shows expression of CD10, CD19, CD22, CD38, CD43, CD79a, CD99, HLA-DR, terminal deoxynucleotidyltransferase, and clonal cytoplasmic immunoglobulin. CD20, CD34, and CD45 expression are variable. CD3, CD14, CD56, and surface immunoglobulin light chain are uncommonly expressed. Surface immunoglobulin light chain expression indicates a more mature B-cell phenotype. Vimentin may be expressed.
Genetic studies show clonal abnormalities, the most frequent being the addition of chromosome 21q material. Translocation (8;22)(q24;q11) has been reported, involving MYC on chromosome 8q24 and IGL@ (immunoglobulin lambda locus) on chromosome 22q11. MYC may also be rearranged in other combinations. IGH@ (immunoglobulin heavy locus) rearrangement is consistently present. T-cell receptor gene rearrangement may also be present.
B-LBL has been reported with other, apparently clonally unrelated hematolymphoid diseases, including idiopathic hypereosinophilic syndrome and mycosis fungoides.
The differential diagnosis includes Burkitt lymphoma’leukemia, diffuse large B-cell lymphoma, the blastic variant of mantle cell lymphoma, T-cell LBL, NK’T cell neoplasms, CD41’CD56 hematodermic neoplasm, myeloid sarcoma (extramedullary myeloid tumor), and nonhematopoietic malignancies. Like B-LBL, T-cell LBL may express CD79a. Also like B-LBL, Ewing sarcoma and primitive neuroectodermal tumor are PAS-positive and express CD99 and vimentin without CD45. Therefore, careful and thorough immunophenotypic evaluation must be performed on suspected cases of B-LBL and small round cell tumor.
TABLE 23.1 WHO 2008 Classification of B-Cell Neoplasms
Extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma)
Nodal marginal zone lymphoma
Follicular lymphoma
Mantle cell lymphoma
Diffuse large B-cell lymphoma (DCBCL), NOS
Mediastinal (thymic) large B-cell lymphoma
Intravascular large B-cell lymphoma
ALK positive DLBCL
Primary effusion lymphoma
Burkitt lymphoma’leukemia
B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma
B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and classical Hodgkin lymphoma
WHO, World Health Organization.
Adapted from WHO.
MATURE B-CELL NEOPLASMS
ALK1-Positive Large B-Cell Lymphoma
ALK1-positive large B-cell lymphoma is a phenotypic variant of large B-cell lymphoma (42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53). It is not considered a separate form of large B-cell lymphoma, but is discussed here to bring attention to its existence and to the differential diagnosis. It accounts for less than 10% of all ALK1-positive lymphomas, the remainder being T-cell and null-cell tumors.
TABLE 23.2 Differential Features of Mature Small B-Cell Neoplasms
B-CLL
LPL
MZL
HCL
FL
MCL
Nucleus
Round
Round
Round
Irregular
Irregular
Irregular
Nucleolus
−
−’+
+
+
+
+
Infiltrate
D,N
D,N,P
D, N, I
D,I
D,N,P
D,N,P,I
CD5
+
−
−
−
−
+
CD10
−
−
−
−
+
−
CD23
+
−
−
+
+’−
−
CD103
−
−
−
+
−
−
FMC7
−’+
+
+
+
+
+
B-CLL, B-cell chronic lymphocytic leukemia’small lymphocytic lymphoma; D, diffuse; FL, follicular lymphoma; HCL, hairy cell leukemia; I, sinusoidal’intravascular; LPL, lymphoplasmacytic lymphoma; MCL, mantle cell lymphoma; MZL, marginal zone lymphoma; N, nodular; P, paratrabecular; +, positive; −, negative; +’−, usually positive, sometimes negative; −’+, usually negative, sometimes positive.
The patients are predominantly adult males, who present with systemic symptoms and disease involving lymph nodes, nasopharynx, liver, stomach, bone marrow, spleen, muscle, and other sites. Lymph node disease is characterized by a sinusoidal and diffuse pattern of infiltration. The clinical course is aggressive.
Laboratory studies may show a serum monoclonal immunoglobulin.
The lymphoma cells have been described as anaplastic, immunoblastic, and plasmacytoid, with large size, scant basophilic cytoplasm, round nucleus, and a single prominent nucleolus.
Peripheral blood is not involved. Bone marrow has rarely been reported as an involved site.
Immunophenotyping shows expression of CD38, CD45, CD138, and clonal cytoplasmic immunoglobulin. ALK1 is usually expressed in a granular cytoplasmic pattern, but may be expressed in a nuclear membrane pattern. CD4, CD20, CD30, CD43, CD57, CD79a, cytokeratin, epithelial membrane antigen (EMA), and perforin may be expressed. CD3, CD56, and TIA-1 are not expressed.
Genetic studies have consistently shown t(2;17)(p23;q23), fusing ALK on chromosome 2p23 to CLTC on chromosome 17q23. Other ALK translocations, including t(2;5)(p23;q35) and t(2;22)(p23;q11.2), have been reported. Immunoglobulin genes are typically rearranged. T-cell receptor genes are usually in germline configuration.
The differential diagnosis includes other tumors expressing ALK1 and EMA, including anaplastic large T-cell and null-cell lymphomas and carcinoma.
B-Cell Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma
B-cell chronic lymphocytic leukemia (B-CLL) and B-cell small lymphocytic lymphoma (B-SLL) are essentially identical, except that B-CLL is leukemic by definition. B-CLL is virtually always accompanied by lymph node or other tissue infiltration, or B-SLL. The reverse is not true. B-SLL is accompanied by B-CLL in approximately 80% of cases. The reason for this discrepancy is not known. In the following discussion, the term B-CLL is used for simplicity to refer to both disorders.
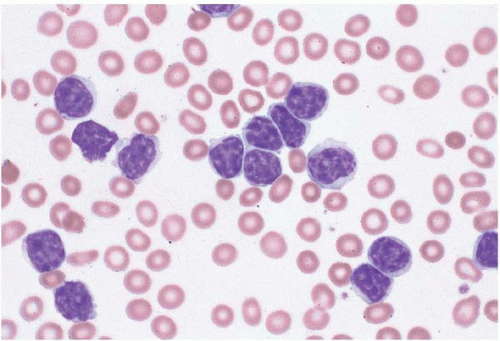
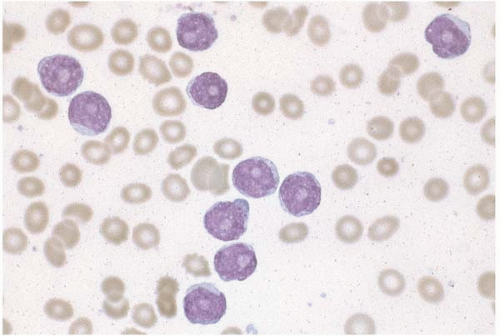
Figure 23.1 B-cell chronic lymphocytic leukemia, peripheral blood. The lymphocytes are small and monomorphous, with round nuclei and condensed chromatin.
Hybrid cases have been reported, showing features of B-CLL and lymphoplasmacytic lymphoma, hairy cell leukemia, follicular lymphoma, mantle cell lymphoma, and multiple myeloma.
Discordant morphology between bone marrow and lymph node biopsies sometimes occurs, with B-CLL in the bone marrow and lymphoplasmacytic lymphoma, marginal zone lymphoma, or diffuse large B-cell lymphoma in other sites.
Figure 23.2 B-cell chronic lymphocytic leukemia in prolymphocytic transformation, peripheral blood. Numerous prolymphocytes are present in this specimen from a patient with longstanding B-cell chronic lymphocytic leukemia.
Figure 23.3 Atypical B-cell chronic lymphocytic leukemia, peripheral blood. The lymphocytes are relatively small and monomorphous but show irregular nuclear contours.
Figure 23.4 Atypical B-cell chronic lymphocytic leukemia, peripheral blood. The lymphocytes are relatively small and monomorphous but show irregular nuclear contours and lobation.
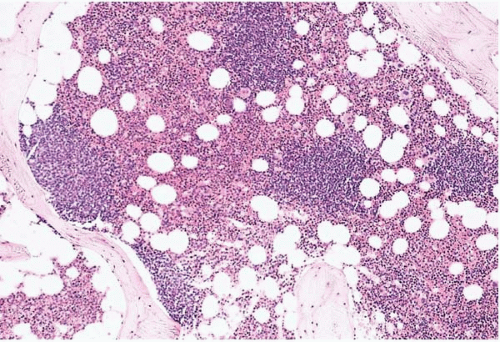
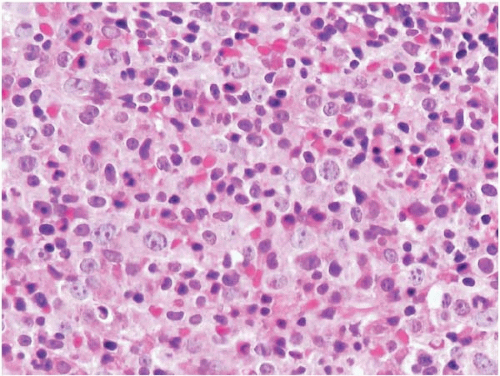
Figure 23.5 B-cell chronic lymphocytic leukemia’small lymphocytic lymphoma, bone marrow biopsy. The hematopoietic tissue is displaced by nodular and interstitial aggregates of clonal B cells.
Figure 23.6 B-cell chronic lymphocytic leukemia’small lymphocytic lymphoma, bone marrow biopsy. A diffuse interstitial infiltrate of small round lymphocytes is present.
Laboratory studies show lymphocytosis and may also show cytopenias, immune-mediated anemia and thrombocytopenia, cold agglutinins, cryoglobulin, serum clonal immunoglobulin, increased serum viscosity, and a neutrophilic leukemoid reaction. Erythrocytosis has been rarely reported.
The absolute lymphocyte count required for the diagnosis of B-CLL is not uniformly established. The widespread use of immunophenotyping by flow cytometry has resulted in the detection of numerous small B-cell clones of undetermined clinical significance in peripheral blood and bone marrow specimens. The general practice is to make a diagnosis of B-CLL with an absolute clonal lymphocyte count of 5.0 × 109‘L or more.
Patients may present with signs and symptoms of cytopenias, lymphadenopathy, hepatosplenomegaly, a mass lesion, and’or Waldenström macroglobulinemia. The course is usually indolent. Spontaneous remission has been reported, sometimes in association with T-cell hyperplasia in the bone marrow.
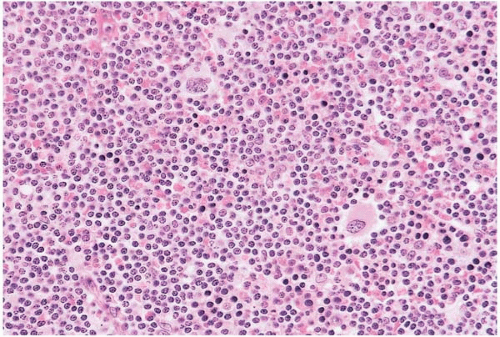
Figure 23.7 B-cell chronic lymphocytic leukemia’small lymphocytic lymphoma, bone marrow biopsy. A poorly defined zone of larger cells is present, constituting a proliferation center.
Peripheral blood and bone marrow aspirate smears show round to ovoid neoplastic lymphocytes that are slightly larger than normal lymphocytes. At low magnification, they may appear triangular, polygonal, or tapered. At high magnification, they show ovoid nuclei with coarsely clumped chromatin and absent or inconspicuous nucleoli. In some cases, the chromatin is quite condensed and shows “cracks” or a cartwheel appearance. The cytoplasm is scanty to moderately abundant, pale blue, and agranular. Atypical cells showing cleaved nuclei or prolymphocytic features may be found.
Disrupted cells are common and usually appear as smudged nuclei devoid of cytoplasm (smudge cells) or ballooned skeins of chromatin (basket cells). Smudge cells are accurately counted as lymphocytes by automated blood analyzers but are routinely excluded from manual white blood cell differential counts, a practice that may lead to underestimation of lymphocytosis in manually reviewed smears.
Histologic sections of the bone marrow show approximately 20% to 90% lymphocytes in nodular, interstitial, diffuse, interfollicular, and’or mixed patterns. The neoplastic cells appear as closely packed, dark-staining nuclei with scant cytoplasm. Scattered larger lymphocytes may be found. Proliferation centers and’or germinal centers may be present. Admixed normal cells include T-cells and dendritic cells.
Other findings include erythroid and’or megakaryocytic hyperplasia owing to immune-mediated cytopenias, pure red cell aplasia, T-cell hyperplasia, reactive mastocytosis, amyloidosis, mu heavy chain disease, and immunoglobulin deposition disease. Evidence of opportunistic infection may be found. Therapy with anti-CD20 (rituximab) may leave residual aggregates of T cells in the bone marrow, which must be distinguished from residual B-CLL.
Interesting findings associated with immunoglobulin production in B-CLL include pseudonuclear immunoglobulin inclusions (Dutcher bodies); multiple spherical bodies formed by immunoglobulin trapped within rough endoplasmic reticulum (Mott cells); single, large, spherical cytoplasmic accumulations of immunoglobulin (Russell bodies); cytoplasmic vacuolation with signet-ring morphology; and immunoglobulin crystals. Intracytoplasmic filamentous inclusions, probably composed of immunoglobulin, have been described. In Mott cells and Russell bodies, the cell nucleus is typically displaced or obscured.
Morphologic variants and atypical forms of B-CLL have been described. The paraimmunoblastic variant is composed of larger cells with more basophilic cytoplasm. A binucleated variant has been reported. Atypical B-CLL is a general term used for cases with unusual clinical, morphologic, immunophenotypic, and’or genetic characteristics. These include: more than 10% prolymphocytes; bright CD20, CD23, and’or surface immunoglobulin light chain expression; clonal karyotypic anomalies; and a more aggressive clinical course. Some cases of atypical B-CLL may be indistinguishable from the leukemic phase of mantle cell lymphoma.
Immunophenotyping usually shows expression of CD5, CD11c, CD19, CD20, CD23, CD43, CD45, HLA-DR, and clonal surface immunoglobulin light chain. CD22, CD79b, and FMC-7 may be expressed. CD38 and ZAP-70 expression have been associated with a relatively poor prognosis. B-CLL may show expression of CD2, CD7, CD8, CD10, CD13, CD14, CD25, CD33, CD34, or CD138, or lack expression of CD5, CD11c, or surface immunoglobulin light chain. Bcl-1 and bcl-2 may be expressed. Weak (dim) CD5 expression must be carefully distinguished from true CD5 negativity because some CD5-negative B-cell neoplasms represent disorders other than B-CLL. Anti-CD20 (rituximab) therapy may eliminate CD20 expression in B-CLL cells. Multiple B-CLL clones occur in some cases, distinguishable by immunophenotype. Minute peripheral blood and bone marrow populations with a B-CLL immunophenotype have been reported; these are of uncertain clinical significance.
Genetic studies show anomalies in more than 80% of cases. These include trisomy 12 and abnormalities involving IGH@ on chromosome 14q32, TP53 on chromosome 17p13, and genes on chromosomes 11q23 and 13q14. TP53 and 11q deletions are indications of clonal evolution and are thus associated with poor survival. Reported translocations include t(8;14)(q24;q32), t(11;14)(q13;q32), and t(14;18)(q32;q21).
Discordance of diagnosis may occur because of sampling and to the different methods used. Bone marrow aspirate, bone marrow biopsy, and flow cytometry may give different results in the same case. In general, flow cytometry appears to be the most sensitive technique for identifying clonal cells in B-CLL.
Transformation to a higher-grade B-cell neoplasm, sometimes called Richter syndrome (RS), occurs in approximately 10% of cases. It is not clear whether RS refers only to clonally related transformation or includes clonally unrelated B-cell tumors as well. Both occur in B-CLL. In prolymphocytic transformation, prolymphocytes exceed 10% of nucleated cells; CD5 and CD23 expression are typically retained. Other apparently clonally related transformations include diffuse large B-cell lymphoma, Hodgkin lymphoma, marginal zone lymphoma, and Burkitt-like, lymphoblastic, plasmacytoid, and histiocytic neoplasms.
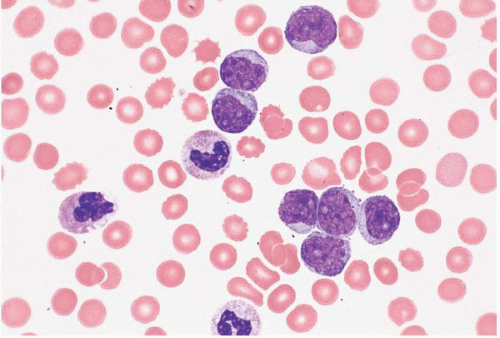
B-CLL has also been reported with apparently clonally unrelated myeloproliferative disorders, myelodysplastic syndromes, acute myeloid leukemia, dendritic cell and histiocytic neoplasms, diffuse large B-cell lymphoma, hairy cell leukemia, mantle cell lymphoma, Hodgkin lymphoma, solitary plasmacytoma, multiple myeloma, anaplastic large cell lymphoma, mycosis fungoides, and other clonal T-cell disorders (Figs. 23.8 and 23.9).
The differential diagnosis includes reactive lymphocytosis, benign lymphoid aggregates, persistent polyclonal lymphocytosis, residual T-cell aggregates following anti-CD20 (rituximab) therapy, hepatitis C-related oligoclonal B-cell proliferation, small clonal B-cell populations of undetermined clinical significance, de novo B-cell prolymphocytic leukemia, lymphoplasmacytic lymphoma, mantle cell lymphoma, marginal zone lymphoma, lymphocyte-predominant Hodgkin lymphoma, and neoplasms composed of small T-cells.
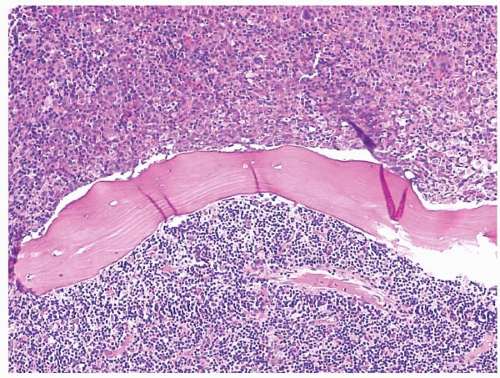
Figure 23.8 B-cell chronic lymphocytic leukemia’small lymphocytic lymphoma and acute myeloid leukemia, bone marrow biopsy. This case of collision tumors shows separate areas of involvement on either side of the bony trabecula. On one side is a relatively homogeneous population of large, eosinophilic, granular and immature myeloid cells. On the other is a homogenous population of small round lymphocytes.
B-Cell Prolymphocytic Leukemia
B-cell prolymphocytic leukemia (B-PLL) occurs de novo and as a transformation of B-cell chronic lymphocytic leukemia (B-CLL), follicular lymphoma, hairy cell leukemia, and mantle cell lymphoma (Figs. 23.10, 23.11and 23.12) (162, 163, 164, 165, 166, 167, 168, 169, 170, 171, 172, 173, 174, 175, 176, 177, 178, 179, 180, 181, 182, 183, 184). B-PLL accounts for 80% of PLL cases, the remainder being of T-cell origin.
Figure 23.9 B-cell chronic lymphocytic leukemia’small lymphocytic lymphoma and acute myeloid leukemia, bone marrow biopsy. At higher power, the area between the two tumors shows a mix of large, eosinophilic myeloid cells and small lymphocytes.
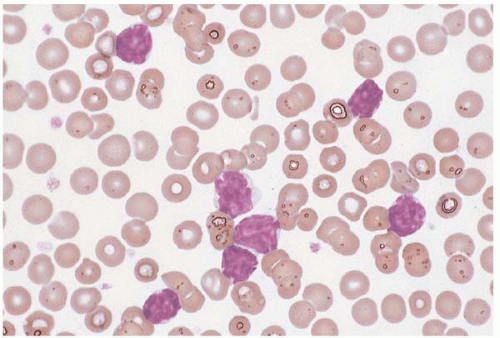
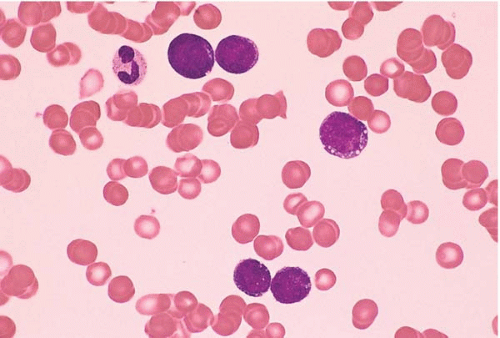
Figure 23.10 B-cell prolymphocytic leukemia, peripheral blood. A monomorphous population of large lymphocytes is present, each cell containing a single large nucleolus.
Laboratory studies show marked leukocytosis, with white blood cell counts often exceeding 100 × 109‘L and prolymphocytes comprising 55% or more of nucleated cells. Anemia, autoimmune hemolytic anemia, and thrombocytopenia may be present.
Patients are usually middle-aged and older adults who present with splenomegaly. Skin and gingival involvement have been reported.
Peripheral blood and marrow smears show prolymphocytes, with large lymphocytes with ovoid nuclei, moderately condensed chromatin, a single prominent, centrally located nucleolus, and moderately abundant pale blue cytoplasm.
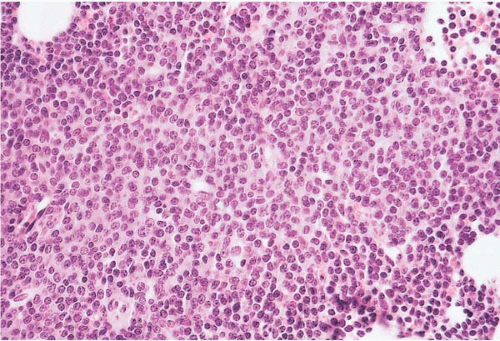
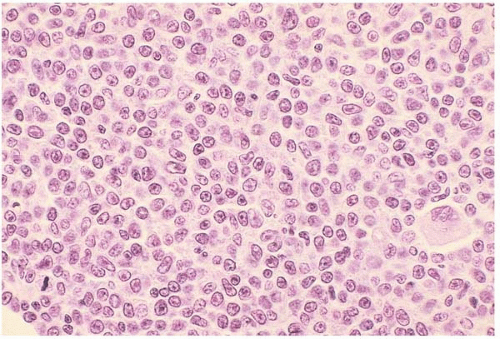
Histologic sections of the bone marrow show an interstitial and’or diffuse infiltrate of prolymphocytes. Reticulin fibrosis may be present.
Figure 23.11 B-cell prolymphocytic leukemia, bone marrow biopsy. A diffuse interstitial infiltrate of monomorphous lymphocytes is present, the majority of cells showing a single nucleolus.
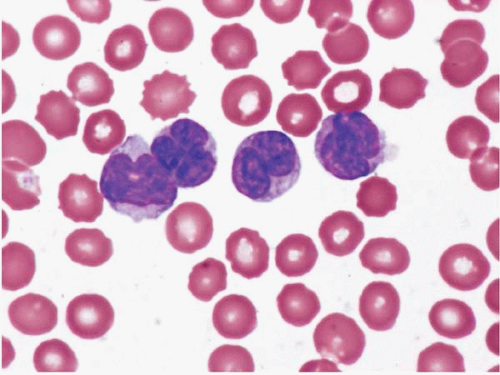
Figure 23.12 Burkitt lymphoma’leukemia, peripheral blood. The lymphocytes are small to medium in size and monomorphous, with evenly dispersed chromatin and scant basophilic cytoplasm containing a few vacuoles.
Immunophenotyping shows expression of CD11c, CD19, CD20, CD22, CD45, CD79b, HLA-DR, and clonal surface immunoglobulin light chain. CD5, CD23, CD38, and FMC7 may be expressed. Occasional cases show CD11b or CD13 expression.
Genetic studies may show trisomy 12 and deletions involving chromosomes 11q23, 13q14, and 17p13, the latter affecting TP53. Also reported are t(8;14)(q24;q32), involving MYC on chromosome 8q24 and IGH@ (immunoglobulin heavy locus) on chromosome 14q32. Other anomalies involving MYC have been described.
B-PLL has been reported with Hodgkin lymphoma and with clonally identical Burkitt lymphoma’leukemia.
The differential diagnosis includes the leukemic phase of large B-cell lymphoma, the nucleolated variant of mantle cell lymphoma, T-cell prolymphocytic leukemia, and acute leukemia. The distinction between de novo B-PLL and prolymphocytic transformation of B-CLL is often blurred, but is not critical because outcomes are similar.
Hybrid cases have been reported, showing features of BLL and precursor B-cell acute lymphoblastic leukemia.
Discordant morphology between bone marrow and lymph node biopsies has been reported, with BLL in the bone marrow and Burkitt-like lymphoma in the lymph node. This distinction is probably not clinically significant.
Laboratory studies may show evidence of Epstein-Barr virus (EBV) infection, lactic acidosis, and increased serum lactate dehydrogenase.
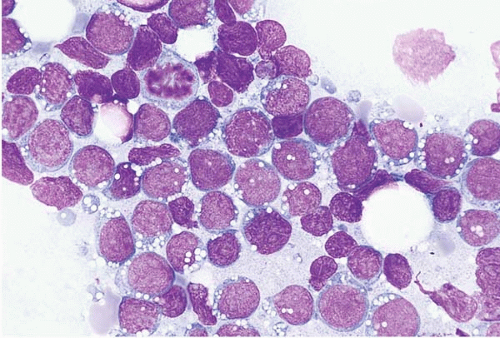
Figure 23.13 Burkitt lymphoma’leukemia, bone marrow aspirate. The malignant cells are large and monomorphous, with round nuclei, dispersed chromatin, and prominently vacuolated basophilic cytoplasm.
Patients may present with a mass and secondary involvement of the peripheral blood and bone marrow, or with leukemia as the primary manifestation and secondary development of a mass. BLL pursues an aggressive clinical course.
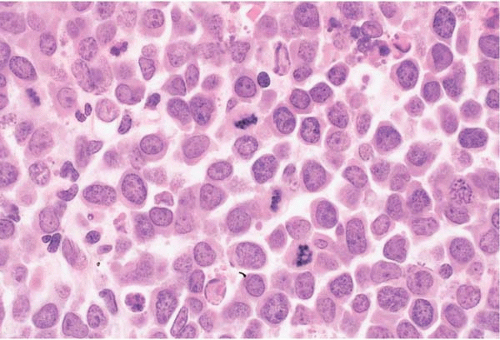
Peripheral blood and bone aspirate smears show medium-sized cells with predominantly round nuclei, multiple small nucleoli, moderately abundant basophilic cytoplasm with prominent vacuolation, and numerous mitoses. The chromatin is dispersed but not as completely or evenly as in blasts.
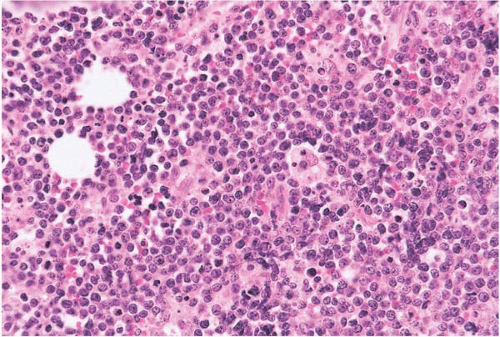
Histologic sections of the bone marrow show a diffuse infiltrate of neoplastic cells with nucleoli and amphophilic cytoplasm. Mitoses are often found. Increased histiocytes may impart a “starry-sky” appearance due to cytoplasmic retraction and may contain ingested nuclear debris. Other findings may include eosinophilic hyperplasia and myelodysplastic changes.
Figure 23.14 Burkitt lymphoma’leukemia, bone marrow biopsy. The hematopoietic tissue is replaced by a diffuse, monomorphous infiltrate with a “starry-sky” pattern, imparted by histiocytes containing ingested cellular remnants.
Figure 23.15 Burkitt lymphoma’leukemia, bone marrow biopsy. The infiltrate is composed of monomorphous cells with round nuclei containing nucleoli and dispersed chromatin; the mitotic rate is high.
Immunophenotyping shows expression of CD10, CD19, CD20, CD22, CD45, FMC-7, HLA-DR, clonal surface immunoglobulin light chain, and bcl-6. Rare cases show expression of CD5 or lack expression of CD20 or surface immunoglobulin light chain.
Genetic studies usually show reciprocal translocation of MYC at chromosome 8q24 and an immunoglobulin locus. The most common abnormality is t(8;14)(q24;q32), involving MYC and IGH@ (immunoglobulin heavy locus), followed by t(8;22)(q24;q11), involving MYC and IGL@ (immunoglobulin lambda locus), and t(2;8)(p12;q24), involving MYC and IGK@ (immunoglobulin kappa locus). Primarily occurring in adult cases is t(14;18)(q32;q21), involving IGH@ and BCL2. Clonal evolution is accompanied by trisomy 12, chromosome 17p anomalies, and TP53 anomalies. Translocation of MYC and an immunoglobulin gene are not pathognomonic of BLL but have also been reported in a variety of B-cell and plasma cell malignancies.
Molecular studies usually show clonal integration of EBV genome into tumor cells.
The differential diagnosis includes acute myeloid leukemia, lymphoblastic malignancy, diffuse large B-cell lymphoma, high-grade B-cell lymphoma, the blastic variant of mantle cell lymphoma, plasma cell leukemia with cytoplasmic vacuolation, and small, round cell tumors of nonhematolymphoid origin.
Only gold members can continue reading. Log In or Register to continue